
De Novo Genome Assembly of the Whitespot Parrotfish (Scarus forsteni): A Valuable Scaridae Genomic Resource
 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and DNA Sequencing
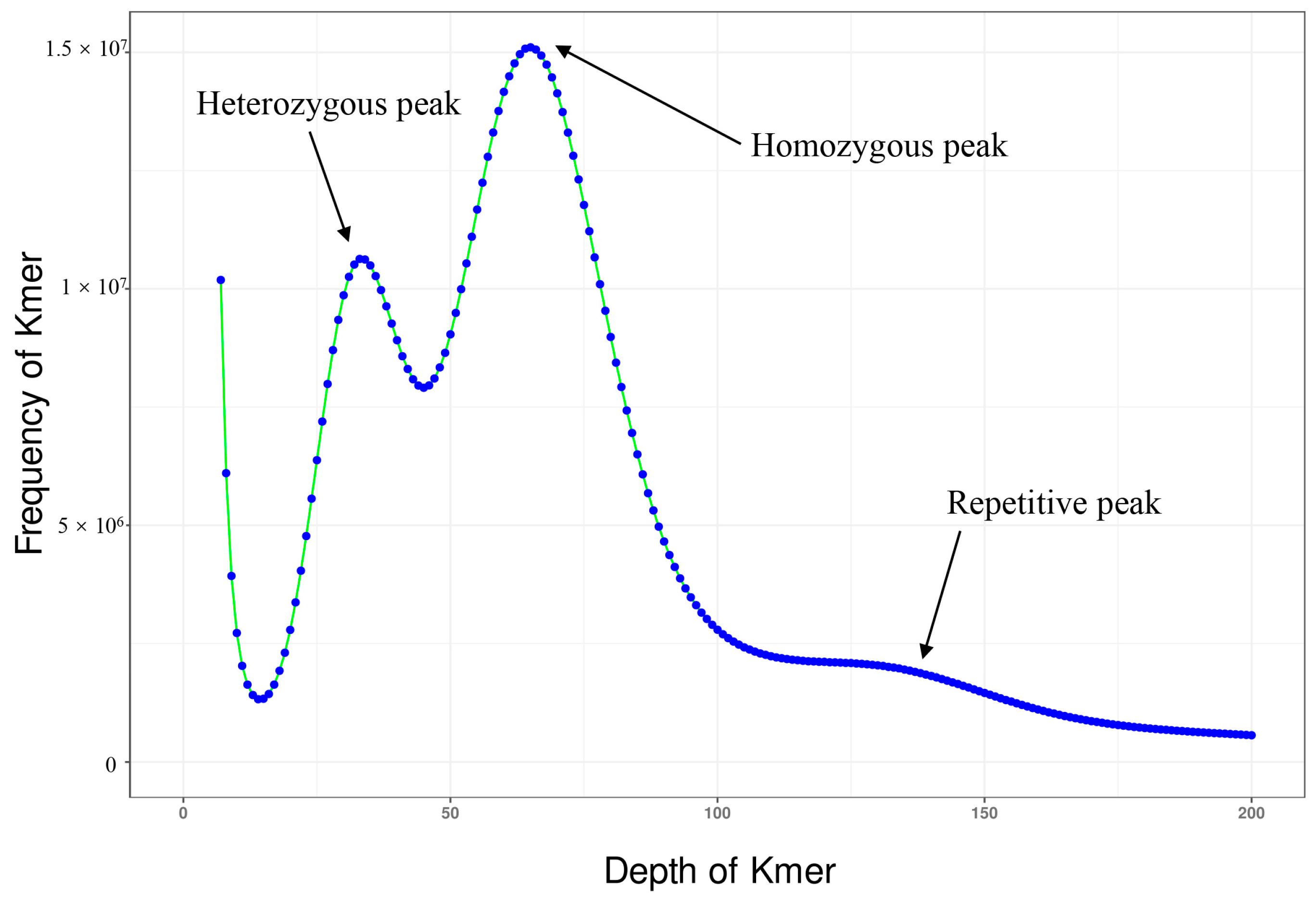
2.2. Sequence Data Processing and Genome Survey
2.3. De Novo Genome Assembly and Quality Assessment
2.4. Gene Prediction and Annotation
2.5. Repetitive Element Annotation
2.6. Population History Analysis
2.7. Phylogenetic Analysis and Divergence Time Estimation
2.8. Expansion and Contraction of Gene Families
2.9. Screening of Positive Selection Genes
3. Results
3.1. Genome Assembly and Quality Assessment
3.2. Genome Annotation
3.3. Repetitive Element Annotation
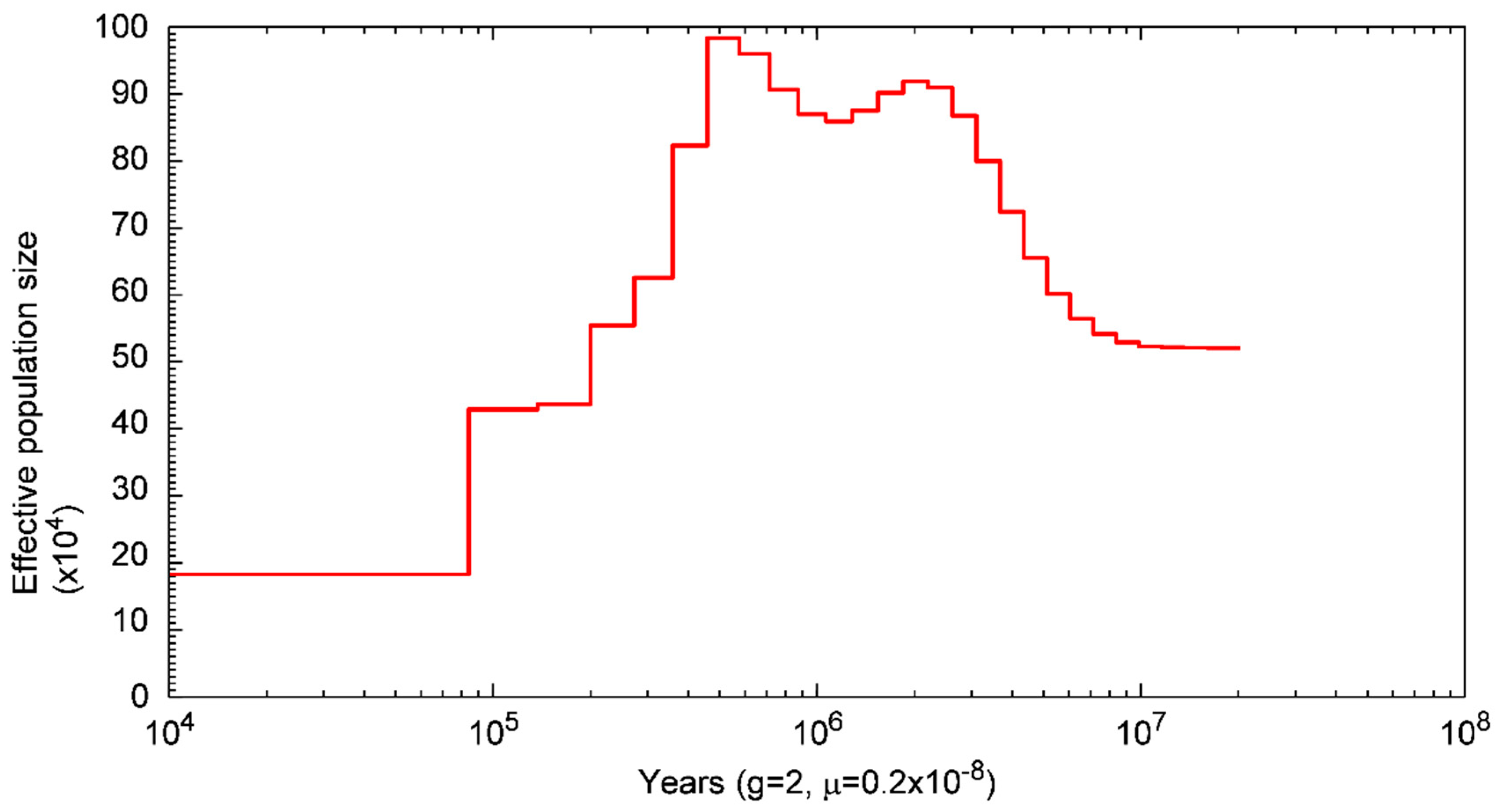
3.4. Population History of S. forsteni
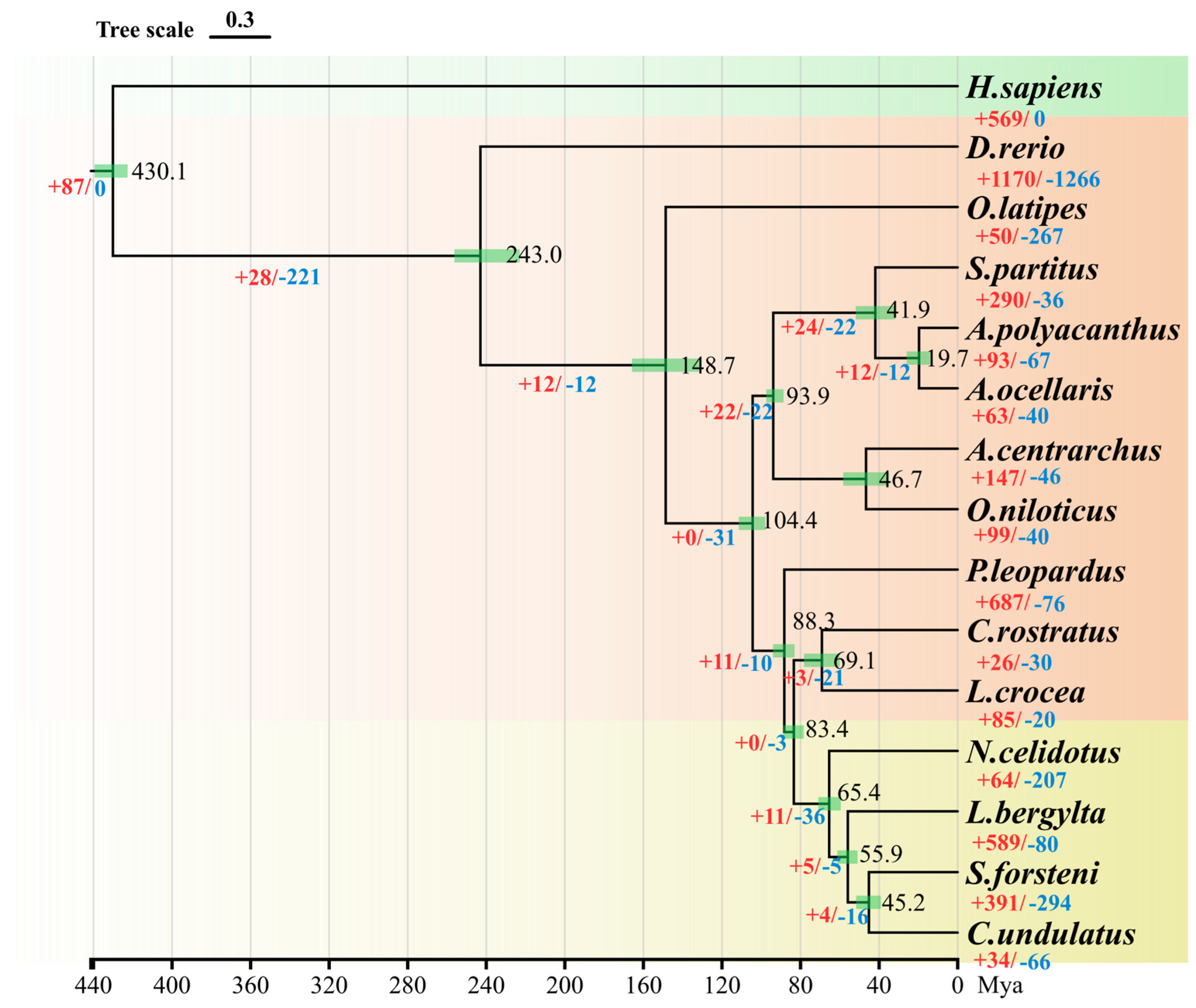
3.5. Genome Evolution Analysis
3.6. Expansion and Contraction of Gene Families
3.7. Analysis of Positive Selection Genes
4. Discussion
4.1. Genome Assembly and Annotation
4.2. Repetitive Element Analysis
4.3. Population History of S. forsteni
4.4. Genome Evolution Analysis
4.5. Enrichment of Expansion and Contraction Gene Families
4.6. Analysis of Positive Selection Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Darius, H.T.; Paillon, C.; Mou-Tham, G.; Ung, A.; Cruchet, P.; Revel, T.; Viallon, J.; Vigliola, L.; Ponton, D.; Chinain, M. Evaluating Age and Growth Relationship to Ciguatoxicity in Five Coral Reef Fish Species from French Polynesia. Mar. Drugs 2022, 20, 251. [Google Scholar] [CrossRef]
- Kazancioglu, E.; Near, T.J.; Hanel, R.; Wainwright, P.C. Influence of Sexual Selection and Feeding Functional Morphology on Diversification Rate of Parrotfishes (Scaridae). Proc. Biol. Sci. 2009, 276, 3439–3446. [Google Scholar] [CrossRef]
- Fitzpatrick, J.M.; Carlon, D.B.; Lippe, C.; Robertson, D.R. The West Pacific Diversity Hotspot as a Source or Sink for New Species? Population Genetic Insights from the Indo-Pacific Parrotfish Scarus rubroviolaceus. Mol. Ecol. 2011, 20, 219–234. [Google Scholar] [CrossRef]
- Gomi, K.; Nakamura, Y.; Kanda, M.; Honda, K.; Nakaoka, M.; Honma, C.; Adachi, M. Diel Vertical Movements and Feeding Behaviour of Blue Humphead Parrotfish Scarus Ovifrons in a Temperate Reef of Japan. J. Fish Biol. 2021, 99, 131–142. [Google Scholar] [CrossRef]
- Carlon, D.B.; Robertson, D.R.; Barron, R.L.; Choat, J.H.; Anderson, D.J.; Schwartz, S.A.; Sánchez-Ortiz, C.A. The Origin of the Parrotfish Species Scarus Compressus in the Tropical Eastern Pacific: Region-Wide Hybridization between Ancient Species Pairs. BMC Ecol. Evol. 2021, 21, 7. [Google Scholar] [CrossRef]
- Cowman, P.F.; Bellwood, D.R.; van Herwerden, L. Dating the Evolutionary Origins of Wrasse Lineages (Labridae) and the Rise of Trophic Novelty on Coral Reefs. Mol. Phylogenet. Evol. 2009, 52, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Li, C.; Yu, D.; Wang, T.; Lin, L.; Xiao, Y.; Wu, P.; Liu, Y. Comparative Mitogenome Analyses Uncover Mitogenome Features and Phylogenetic Implications of the Parrotfishes (Perciformes: Scaridae). Biology 2023, 12, 410. [Google Scholar] [CrossRef] [PubMed]
- Alfaro, M.E.; Brock, C.D.; Banbury, B.L.; Wainwright, P.C. Does Evolutionary Innovation in Pharyngeal Jaws Lead to Rapid Lineage Diversification in Labrid Fishes? BMC Evol. Biol. 2009, 9, 255. [Google Scholar] [CrossRef] [PubMed]
- Streelman, J.T.; Alfaro, M.; Westneat, M.W.; Bellwood, D.R.; Karl, S.A. Evolutionary History of the Parrotfishes: Biogeography, Ecomorphology, and Comparative Diversity. Evol. Int. J. Org. Evol. 2002, 56, 961–971. [Google Scholar] [CrossRef]
- Nanami, A. Parrotfish Grazing Ability: Interspecific Differences in Relation to Jaw-Lever Mechanics and Relative Weight of Adductor Mandibulae on an Okinawan Coral Reef. PeerJ 2016, 4, e2425. [Google Scholar] [CrossRef]
- Pereira, P.H.C.; Santos, M.; Lippi, D.L.; Silva, P. Ontogenetic Foraging Activity and Feeding Selectivity of the Brazilian Endemic Parrotfish Scarus zelindae. PeerJ 2016, 4, e2536. [Google Scholar] [CrossRef] [PubMed]
- Carr, A.; Kemp, A.; Tibbetts, I.; Truss, R.; Drennan, J. Microstructure of Pharyngeal Tooth Enameloid in the Parrotfish Scarus rivulatus (Pisces: Scaridae). J. Microsc. 2006, 221, 8–16. [Google Scholar] [CrossRef]
- Viviani, J.; LeBlanc, A.; Rurua, V.; Mou, T.; Liao, V.; Lecchini, D.; Galzin, R.; Viriot, L. Plicidentine in the Oral Fangs of Parrotfish (Scarinae, Labriformes). J. Anat. 2022, 241, 601–615. [Google Scholar] [CrossRef]
- Goldberg, E.G.; Raab, T.K.; Desalles, P.; Briggs, A.A.; Dunbar, R.B.; Millero, F.J.; Woosley, R.J.; Young, H.S.; Micheli, F.; Mccauley, D.J. Chemistry of the Consumption and Excretion of the Bumphead Parrotfish (Bolbometopon muricatum), a Coral Reef Mega-Consumer. Coral Reefs 2019, 38, 347–357. [Google Scholar] [CrossRef]
- Abdel-Aziz, E.-S.H.; Bawazeer, F.A.; El-Sayed Ali, T.; Al-Otaibi, M. Sexual Patterns and Protogynous Sex Reversal in the Rusty Parrotfish, Scarus ferrugineus (Scaridae): Histological and Physiological Studies. Fish Physiol. Biochem. 2012, 38, 1211–1224. [Google Scholar] [CrossRef]
- Godwin, J.; Sawby, R.; Warner, R.R.; Crews, D.; Grober, M.S. Hypothalamic Arginine Vasotocin mRNA Abundance Variation across Sexes and with Sex Change in a Coral Reef Fish. Brain. Behav. Evol. 2000, 55, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Andradi-Brown, D.A.; Gress, E.; Wright, G.; Exton, D.A.; Rogers, A.D. Reef Fish Community Biomass and Trophic Structure Changes across Shallow to Upper-Mesophotic Reefs in the Mesoamerican Barrier Reef, Caribbean. PLoS ONE 2016, 11, e0156641. [Google Scholar] [CrossRef]
- Nanami, A. Spatial Distribution of Parrotfishes and Groupers in an Okinawan Coral Reef: Size-Related Associations in Relation to Habitat Characteristics. PeerJ 2021, 9, e12134. [Google Scholar] [CrossRef]
- Guo, W.; Bokade, R.; Cohen, A.L.; Mollica, N.R.; Leung, M.; Brainard, R.E. Ocean Acidification Has Impacted Coral Growth on the Great Barrier Reef. Geophys. Res. Lett. 2020, 47, e2019GL086761. [Google Scholar] [CrossRef]
- Burkepile, D.E.; Hay, M.E. Impact of Herbivore Identity on Algal Succession and Coral Growth on a Caribbean Reef. PLoS ONE 2010, 5, e8963. [Google Scholar] [CrossRef]
- Bezerra, I.M.; Gramacho, K.P.; Barreto, M.A.; Hackradt, C.W.; Leão Feitosa, J.L.; Torres, R.A.; Ferreira, B.P.; González-Wanguemert, M.; Félix-Hackradt, F.C. Genetic Diversity and Gene Flow of the Threatened Brazilian Endemic Parrotfish Scarus trispinosus (Valenciennes, 1840). Mar. Environ. Res. 2018, 142, 155–162. [Google Scholar] [CrossRef]
- Labrador, K.; Fortaleza, M.; Cabasan, J.; Elumba, M.; Nañola, C. Genetic Diversity and Population Connectivity of the Greenblotch Parrotfish (Scarus quoyi, Valenciennes, 1840) within Southern Mindanao Inferred from Mitochondrial 16S rRNA. Philipp. J. Sci. 2022, 151, 2305–2310. [Google Scholar] [CrossRef]
- Pereira, P.H.C.; Araujo, J.C.; Lima, G.V.; Côrtes, L.G.F.; Gomes, E.; Magris, R.A. Effectiveness of Management Zones for Recovering Parrotfish Species within the Largest Coastal Marine Protected Area in Brazil. Sci. Rep. 2022, 12, 12232. [Google Scholar] [CrossRef]
- Smith, L.L.; Fessler, J.L.; Alfaro, M.E.; Streelman, J.T.; Westneat, M.W. Phylogenetic Relationships and the Evolution of Regulatory Gene Sequences in the Parrotfishes. Mol. Phylogenet. Evol. 2008, 49, 136–152. [Google Scholar] [CrossRef]
- Austin, C.M.; Tan, M.H.; Harrisson, K.A.; Lee, Y.P.; Croft, L.J.; Sunnucks, P.; Pavlova, A.; Gan, H.M. De Novo Genome Assembly and Annotation of Australia’s Largest Freshwater Fish, the Murray Cod (Maccullochella peelii), from Illumina and Nanopore Sequencing Read. GigaScience 2017, 6, gix063. [Google Scholar] [CrossRef]
- Fernandez-Silva, I.; Henderson, J.B.; Rocha, L.A.; Simison, W.B. Whole-Genome Assembly of the Coral Reef Pearlscale Pygmy Angelfish (Centropyge vrolikii). Sci. Rep. 2018, 8, 1498. [Google Scholar] [CrossRef]
- Paris, J.R.; Stevens, J.R.; Catchen, J.M. Lost in Parameter Space: A Road Map for Stacks. Methods Ecol. Evol. 2017, 8, 1360–1373. [Google Scholar] [CrossRef]
- Galla, S.J.; Forsdick, N.J.; Brown, L.; Hoeppner, M.P.; Knapp, M.; Maloney, R.F.; Moraga, R.; Santure, A.W.; Steeves, T.E. Reference Genomes from Distantly Related Species Can Be Used for Discovery of Single Nucleotide Polymorphisms to Inform Conservation Management. Genes 2019, 10, 9. [Google Scholar] [CrossRef] [PubMed]
- Bariche, M.; Bernardi, G. Lack of a Genetic Bottleneck in a Recent Lessepsian Bioinvader, the Blue-Barred Parrotfish, Scarus ghobban. Mol. Phylogenet. Evol. 2009, 53, 592–595. [Google Scholar] [CrossRef]
- Jawad, L.A. A Comparative Morphological Investigation of Otoliths of Six Parrotfish Species (Scaridae) from the Solomon Islands. J. Fish Biol. 2018, 93, 1046–1058. [Google Scholar] [CrossRef]
- Guo, L.; Zhang, N.; Zhu, K.-C.; Guo, H.-Y.; Liu, B.-S.; Zhang, D.-C. The Complete Mitochondrial Genome of Cheilinus oxycephalus (Perciformes: Labridae). Mitochondrial DNA Part B Resour. 2019, 4, 3816–3817. [Google Scholar] [CrossRef] [PubMed]
- Wingett, S.W.; Andrews, S. FastQ Screen: A Tool for Multi-Genome Mapping and Quality Control. F1000Research 2018, 7, 1338. [Google Scholar] [CrossRef]
- Xiao, Y.; Xiao, Z.; Ma, D.; Liu, J.; Li, J. Genome Sequence of the Barred Knifejaw Oplegnathus fasciatus (Temminck & Schlegel, 1844): The First Chromosome-Level Draft Genome in the Family Oplegnathidae. GigaScience 2019, 8, giz013. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, Y.; Shi, C.; Huang, Z.; Zhang, Y.; Li, S.; Li, Y.; Ye, J.; Yu, C.; Li, Z.; et al. SOAPnuke: A MapReduce Acceleration-Supported Software for Integrated Quality Control and Preprocessing of High-Throughput Sequencing Data. GigaScience 2018, 7, gix120. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Shi, Y.; Yuan, J.; Hu, X.; Zhang, H.; Li, N.; Li, Z.; Chen, Y.; Mu, D.; Fan, W. Estimation of Genomic Characteristics by Analyzing K-Mer Frequency in de Novo Genome Projects. Quant. Biol. 2020, arXiv:1308.2012v2. [Google Scholar] [CrossRef]
- Hu, J.; Fan, J.; Sun, Z.; Liu, S. NextPolish: A Fast and Efficient Genome Polishing Tool for Long-Read Assembly. Bioinformatics 2020, 36, 2253–2255. [Google Scholar] [CrossRef]
- Li, H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. Quant. Biol. 2013, arXiv:1303.3997v2. [Google Scholar] [CrossRef]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Simão, F.A.; Zdobnov, E.M. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Mol. Biol. Evol. 2021, 38, 4647–4654. [Google Scholar] [CrossRef]
- Flynn, J.M.; Hubley, R.; Goubert, C.; Rosen, J.; Clark, A.G.; Feschotte, C.; Smit, A.F. RepeatModeler2 for Automated Genomic Discovery of Transposable Element Families. Proc. Natl. Acad. Sci. USA 2020, 117, 9451–9457. [Google Scholar] [CrossRef]
- Tarailo-Graovac, M.; Chen, N. Using RepeatMasker to Identify Repetitive Elements in Genomic Sequences. Curr. Protoc. Bioinform. 2009, 4.10.1–4.10.14. [Google Scholar] [CrossRef]
- Stanke, M.; Keller, O.; Gunduz, I.; Hayes, A.; Waack, S.; Morgenstern, B. AUGUSTUS: Ab Initio Prediction of Alternative Transcripts. Nucleic Acids Res. 2006, 34, W435–W439. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Salzberg, S.L.; Zhu, W.; Pertea, M.; Allen, J.E.; Orvis, J.; White, O.; Buell, C.R.; Wortman, J.R. Automated Eukaryotic Gene Structure Annotation Using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008, 9, R7. [Google Scholar] [CrossRef] [PubMed]
- Birney, E.; Clamp, M.; Durbin, R. GeneWise and Genomewise. Genome Res. 2004, 14, 988–995. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Guo, H.-B.; Jia, L.; Deng, Q.-X.; Zhu, C.-H.; Zhang, B. High-Quality Chromosome-Level Genome Assembly of Redlip Mullet (Planiliza haematocheila). Zool. Res. 2021, 42, 796–799. [Google Scholar] [CrossRef] [PubMed]
- UniProt Consortium. The Universal Protein Resource (UniProt) 2009. Nucleic Acids Res. 2009, 37, D169–D174. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Zdobnov, E.M.; Apweiler, R. InterProScan—An Integration Platform for the Signature-Recognition Methods in InterPro. Bioinformatics 2001, 17, 847–848. [Google Scholar] [CrossRef] [PubMed]
- Nawrocki, E.P. Annotating Functional RNAs in Genomes Using Infernal. Methods Mol. Biol. 2014, 1097, 163–197. [Google Scholar] [CrossRef]
- Xu, Z.; Wang, H. LTR_FINDER: An Efficient Tool for the Prediction of Full-Length LTR Retrotransposons. Nucleic Acids Res. 2007, 35, W265–W268. [Google Scholar] [CrossRef]
- Benson, G. Tandem Repeats Finder: A Program to Analyze DNA Sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef]
- Chalopin, D.; Naville, M.; Plard, F.; Galiana, D.; Volff, J.-N. Comparative Analysis of Transposable Elements Highlights Mobilome Diversity and Evolution in Vertebrates. Genome Biol. Evol. 2015, 7, 567–580. [Google Scholar] [CrossRef]
- Conte, M.A.; Gammerdinger, W.J.; Bartie, K.L.; Penman, D.J.; Kocher, T.D. A High Quality Assembly of the Nile Tilapia (Oreochromis niloticus) Genome Reveals the Structure of Two Sex Determination Regions. BMC Genom. 2017, 18, 341. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Shen, D.; Xue, S.; Chen, C.; Cui, H.; Song, C. The Contribution of Transposable Elements to Size Variations between Four Teleost Genomes. Mob. DNA 2016, 7, 4. [Google Scholar] [CrossRef] [PubMed]
- Ryu, T.; Herrera, M.; Moore, B.; Izumiyama, M.; Kawai, E.; Laudet, V.; Ravasi, T. A Chromosome-Scale Genome Assembly of the False Clownfish, Amphiprion ocellaris. G3 2022, 12, jkac074. [Google Scholar] [CrossRef] [PubMed]
- Shao, F.; Han, M.; Peng, Z. Evolution and Diversity of Transposable Elements in Fish Genomes. Sci. Rep. 2019, 9, 15399. [Google Scholar] [CrossRef] [PubMed]
- Mather, N.; Traves, S.M.; Ho, S.Y.W. A Practical Introduction to Sequentially Markovian Coalescent Methods for Estimating Demographic History from Genomic Data. Ecol. Evol. 2020, 10, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve Years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.D.; Rivera Hernández, J.M.; Shervette, V.R. Princess Parrotfish Scarus Taeniopterus Age, Growth, Maturity, and Transition. Environ. Biol. Fishes 2021, 104, 581–595. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic Orthology Inference for Comparative Genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML Version 8: A Tool for Phylogenetic Analysis and Post-Analysis of Large Phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Mendes, F.K.; Vanderpool, D.; Fulton, B.; Hahn, M.W. CAFE 5 Models Variation in Evolutionary Rates among Gene Families. Bioinformatics 2021, 36, 5516–5518. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R Package for Comparing Biological Themes among Gene Clusters. Omics J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Gao, F.; Chen, C.; Arab, D.A.; Du, Z.; He, Y.; Ho, S.Y.W. EasyCodeML: A Visual Tool for Analysis of Selection Using CodeML. Ecol. Evol. 2019, 9, 3891–3898. [Google Scholar] [CrossRef]
- Zeisel, A.; Zuk, O.; Domany, E. FDR Control with Adaptive Procedures and FDR Monotonicity. Ann. Appl. Stat. 2011, 5, 943–968. [Google Scholar] [CrossRef]
- Cantalapiedra, C.P.; Hernández-Plaza, A.; Letunic, I.; Bork, P.; Huerta-Cepas, J. eggNOG-Mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Mol. Biol. Evol. 2021, 38, 5825–5829. [Google Scholar] [CrossRef]
- McCartney, A.M.; Hilario, E.; Choi, S.-S.; Guhlin, J.; Prebble, J.M.; Houliston, G.; Buckley, T.R.; Chagné, D. An Exploration of Assembly Strategies and Quality Metrics on the Accuracy of the Rewarewa (Knightia excelsa) Genome. Mol. Ecol. Resour. 2021, 21, 2125–2144. [Google Scholar] [CrossRef]
- Sims, D.; Sudbery, I.; Ilott, N.E.; Heger, A.; Ponting, C.P. Sequencing Depth and Coverage: Key Considerations in Genomic Analyses. Nat. Rev. Genet. 2014, 15, 121–132. [Google Scholar] [CrossRef]
- Liu, D.; Wang, X.; Guo, H.; Zhang, X.; Zhang, M.; Tang, W. Chromosome-Level Genome Assembly of the Endangered Humphead Wrasse Cheilinus undulatus: Insight into the Expansion of Opsin Genes in Fishes. Mol. Ecol. Resour. 2021, 21, 2388–2406. [Google Scholar] [CrossRef]
- Tshilate, T.S.; Ishengoma, E.; Rhode, C. A First Annotated Genome Sequence for Haliotis Midae with Genomic Insights into Abalone Evolution and Traits of Economic Importance. Mar. Genom. 2023, 70, 101044. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Zhang, X.; Wang, X.; Li, J.; Liu, G.; Kuang, Y.; Xu, J.; Zheng, X.; Ren, L.; Wang, G.; et al. Genome Sequence and Genetic Diversity of the Common Carp, Cyprinus carpio. Nat. Genet. 2014, 46, 1212–1219. [Google Scholar] [CrossRef]
- Mattingsdal, M.; Jentoft, S.; Tørresen, O.K.; Knutsen, H.; Hansen, M.M.; Robalo, J.I.; Zagrodzka, Z.; André, C.; Gonzalez, E.B. A Continuous Genome Assembly of the Corkwing Wrasse (Symphodus melops). Genomics 2018, 110, 399–403. [Google Scholar] [CrossRef]
- Bernardi, G.; DeBiasse, M.; Escalona, M.; Marimuthu, M.P.A.; Nguyen, O.; Sacco, S.; Beraut, E.; Miller, C.; Toffelmier, E.; Shaffer, H.B. Reference Genome of the California Sheephead, Semicossyphus pulcher (Labridae, Perciformes), A Keystone Fish Predator in Kelp Forest Ecosystems. J. Hered. 2022, 113, 649–656. [Google Scholar] [CrossRef]
- Kang, J.; Ramirez-Calero, S.; Paula, J.R.; Chen, Y.; Schunter, C. Gene Losses, Parallel Evolution and Heightened Expression Confer Adaptations to Dedicated Cleaning Behaviour. BMC Biol. 2023, 21, 180. [Google Scholar] [CrossRef]
- Symonová, R.; Suh, A. Nucleotide Composition of Transposable Elements Likely Contributes to AT/GC Compositional Homogeneity of Teleost Fish Genomes. Mob. DNA 2019, 10, 49. [Google Scholar] [CrossRef] [PubMed]
- Mäkinen, V.; Salmela, L.; Ylinen, J. Normalized N50 Assembly Metric Using Gap-Restricted Co-Linear Chaining. BMC Bioinform. 2012, 13, 255. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-H.; Xu, M.-R.-X.; Wang, P.-L.; Zhu, Z.-G.; Nie, C.-F.; Xiong, X.-M.; Wang, L.; Xie, Z.-Z.; Wen, X.; Zeng, Q.-X.; et al. High-Quality Genome Assembly and Transcriptome of Ancherythroculter nigrocauda, an Endemic Chinese Cyprinid Species. Mol. Ecol. Resour. 2020, 20, 882–891. [Google Scholar] [CrossRef]
- Kidwell, M.G. Transposable Elements and the Evolution of Genome Size in Eukaryotes. Genetica 2002, 115, 49–63. [Google Scholar] [CrossRef]
- Kushwaha, B.; Nagpure, N.S.; Srivastava, S.; Pandey, M.; Kumar, R.; Raizada, S.; Agarwal, S.; Singh, M.; Basheer, V.S.; Kumar, R.G.; et al. Genome Size Estimation and Its Associations with Body Length, Chromosome Number and Evolution in Teleost Fishes. Gene 2023, 864, 147294. [Google Scholar] [CrossRef]
- Canapa, A.; Barucca, M.; Biscotti, M.A.; Forconi, M.; Olmo, E. Transposons, Genome Size, and Evolutionary Insights in Animals. Cytogenet. Genome Res. 2015, 147, 217–239. [Google Scholar] [CrossRef] [PubMed]
- Frantz, L.A.F.; Mullin, V.E.; Pionnier-Capitan, M.; Lebrasseur, O.; Ollivier, M.; Perri, A.; Linderholm, A.; Mattiangeli, V.; Teasdale, M.D.; Dimopoulos, E.A.; et al. Genomic and Archaeological Evidence Suggest a Dual Origin of Domestic Dogs. Science 2016, 352, 1228–1231. [Google Scholar] [CrossRef]
- Johnson, R.N.; O’Meally, D.; Chen, Z.; Etherington, G.J.; Ho, S.Y.W.; Nash, W.J.; Grueber, C.E.; Cheng, Y.; Whittington, C.M.; Dennison, S.; et al. Adaptation and Conservation Insights from the Koala Genome. Nat. Genet. 2018, 50, 1102–1111. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.; Schuster, S.C.; Welch, A.J.; Ratan, A.; Bedoya-Reina, O.C.; Zhao, F.; Kim, H.L.; Burhans, R.C.; Drautz, D.I.; Wittekindt, N.E.; et al. Polar and Brown Bear Genomes Reveal Ancient Admixture and Demographic Footprints of Past Climate Change. Proc. Natl. Acad. Sci. USA 2012, 109, E2382–E2390. [Google Scholar] [CrossRef]
- Nadachowska-Brzyska, K.; Burri, R.; Olason, P.I.; Kawakami, T.; Smeds, L.; Ellegren, H. Demographic Divergence History of Pied Flycatcher and Collared Flycatcher Inferred from Whole-Genome Re-Sequencing Data. PLoS Genet. 2013, 9, e1003942. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Zhao, J.; Li, C. High-Quality Genome Assembly and Annotation of the Big-Eye Mandarin Fish (Siniperca knerii). G3 GenesGenomesGenetics 2020, 10, 877–880. [Google Scholar] [CrossRef]
- Barash, M.S. Environmental Changes in the Neogene and the Biotic Response. Oceanology 2011, 51, 306–314. [Google Scholar] [CrossRef]
- Arias, C.F.; Dikow, R.B.; McMillan, W.O.; De León, L.F. De Novo Genome Assembly of the Electric Fish Brachyhypopomus Occidentalis (Hypopomidae, Gymnotiformes). Genome Biol. Evol. 2021, 13, evab223. [Google Scholar] [CrossRef]
- Hayward, B.; Kawagata, S.; Grenfell, H.; Sabaa, A.; O’Neill, T. Last Global Extinction in the Deep Sea during the Mid-Pleistocene Climate Transition. Paleoceanography 2007, 22. [Google Scholar] [CrossRef]
- Modys, A.B.; Oleinik, A.; Mortlock, R.A.; Toth, L.T.; Precht, W.F. Climate-Modulated Range Expansion of Reef-Building Coral Communities off Southeast Florida during the Late Holocene. Front. Mar. Sci. 2022, 9, 995256. [Google Scholar] [CrossRef]
- Li, J.; Bian, C.; Yi, Y.; Yu, H.; You, X.; Shi, Q. Temporal Dynamics of Teleost Populations during the Pleistocene: A Report from Publicly Available Genome Data. BMC Genom. 2021, 22, 490. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.Z.; Jijun, L.; Zhang, S.Q.; Zhao, J.D.; Cui, J.X. Quaternary Glaciations in China. In Developments in Quaternary Sciences; Ehlers, J., Gibbard, P.L., Eds.; Quaternary Glaciations Extent and Chronology; Elsevier: Amsterdam, The Netherlands, 2004; Volume 2, pp. 105–113. [Google Scholar]
- Westneat, M.W.; Alfaro, M.E.; Wainwright, P.C.; Bellwood, D.R.; Grubich, J.R.; Fessler, J.L.; Clements, K.D.; Smith, L.L. Local Phylogenetic Divergence and Global Evolutionary Convergence of Skull Function in Reef Fishes of the Family Labridae. Proc. Biol. Sci. 2005, 272, 993–1000. [Google Scholar] [CrossRef] [PubMed]
- Westneat, M.W.; Alfaro, M.E. Phylogenetic Relationships and Evolutionary History of the Reef Fish Family Labridae. Mol. Phylogenet. Evol. 2005, 36, 370–390. [Google Scholar] [CrossRef] [PubMed]
- Hughes, L.C.; Nash, C.M.; White, W.T.; Westneat, M.W. Concordance and Discordance in the Phylogenomics of the Wrasses and Parrotfishes (Teleostei: Labridae). Syst. Biol. 2023, 72, 530–543. [Google Scholar] [CrossRef] [PubMed]
- Roff, G.; Ledlie, M.H.; Ortiz, J.C.; Mumby, P.J. Spatial Patterns of Parrotfish Corallivory in the Caribbean: The Importance of Coral Taxa, Density and Size. PLoS ONE 2011, 6, e29133. [Google Scholar] [CrossRef] [PubMed]
- Denny, C.; Schiel, D. Feeding Ecology of the Banded Wrasse Notolabrus fucicola (Labridae) in Southern New Zealand. N. Z. J. Mar. Freshw. Res. 2001, 35, 925–933. [Google Scholar] [CrossRef]
- Greenwood, P.H.; Rosen, D.E.; Weitzman, S.H.; Myers, G.S. George S. Phyletic Studies of Teleostean Fishes, with a Provisional Classification of Living Forms. Bull. AMNH 1966, 131, 4. [Google Scholar]
- Black, A.; Imhoff, V.; Leese, J.; Weimann, S.; Gumm, J.; Richter, M.; Itzkowitz, M. Attack Intensity by Two Species of Territorial Damselfish (Pomacentridae) as Estimates of Competitive Overlap with Two Species of Wrasse (Labridae). J. Ethol. 2014, 32, 63–68. [Google Scholar] [CrossRef]
- Mikami, Y. Phylogenic Relationship of Labridae Species Deduced from Comparative Dissection. Anat. Rec. 2013, 296, 788–797. [Google Scholar] [CrossRef]
- Liang, Y.; Pan, J.-M.; Zhu, K.-C.; Xian, L.; Guo, H.-Y.; Liu, B.-S.; Zhang, N.; Yang, J.-W.; Zhang, D.-C. Genome-Wide Identification of Trachinotus Ovatus Antimicrobial Peptides and Their Immune Response against Two Pathogen Challenges. Mar. Drugs 2023, 21, 505. [Google Scholar] [CrossRef]
- Parkin, J.; Cohen, B. An Overview of the Immune System. Lancet 2001, 357, 1777–1789. [Google Scholar] [CrossRef] [PubMed]
- Arai, T.; Amalina, R.; Bachok, Z. Fatty Acid Composition Indicating Diverse Habitat Use in Coral Reef Fishes in the Malaysian South China Sea. Biol. Res. 2015, 48, 13. [Google Scholar] [CrossRef] [PubMed]
- Sukumaran, S.; Sebastian, W.; Gopalakrishnan, A.; Mathew, O.K.; Vysakh, V.G.; Rohit, P.; Jena, J.K. The Sequence and de Novo Assembly of the Genome of the Indian Oil Sardine, Sardinella longiceps. Sci. Data 2023, 10, 565. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, G.; Atema, J.; Kingsford, M.J.; Black, K.P.; Miller-Sims, V. Smelling Home Can Prevent Dispersal of Reef Fish Larvae. Proc. Natl. Acad. Sci. USA 2007, 104, 858–863. [Google Scholar] [CrossRef]
- Zhang, S.; Song, Y.; Liu, M.; Yuan, Z.; Zhang, M.; Zhang, H.; Seim, I.; Fan, G.; Liu, S.; Liu, X. Chromosome-Level Genome of Butterflyfish Unveils Genomic Features of Unique Colour Patterns and Morphological Traits. DNA Res. Int. J. Rapid Publ. Rep. Genes Genomes 2023, 30, dsad018. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Li, L.; Lv, L.-Y.; Cai, W.-J.; Dou, Y.-Q.; Li, J.; Tang, S.-L.; Chen, X.; Zhang, Z.; Xu, J.; et al. Mandarin Fish (Sinipercidae) Genomes Provide Insights into Innate Predatory Feeding. Commun. Biol. 2020, 3, 361. [Google Scholar] [CrossRef]
- Bao, L.; Tian, C.; Liu, S.; Zhang, Y.; Elaswad, A.; Yuan, Z.; Khalil, K.; Sun, F.; Yang, Y.; Zhou, T.; et al. The Y Chromosome Sequence of the Channel Catfish Suggests Novel Sex Determination Mechanisms in Teleost Fish. BMC Biol. 2019, 17, 6. [Google Scholar] [CrossRef]
- Todd, E.V.; Ortega-Recalde, O.; Liu, H.; Lamm, M.S.; Rutherford, K.M.; Cross, H.; Black, M.A.; Kardailsky, O.; Marshall Graves, J.A.; Hore, T.A.; et al. Stress, Novel Sex Genes, and Epigenetic Reprogramming Orchestrate Socially Controlled Sex Change. Sci. Adv. 2019, 5, eaaw7006. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Item | Value |
|---|---|
| Total length (bp) | 1,713,752,192 |
| Total contigs | 544 |
| GC (%) | 39.32 |
| N50 (bp) | 17,972,633 |
| N90 (bp) | 1,420,458 |
| Ave length (bp) | 3,150,279 |
| Max length (bp) | 58,576,118 |
| Min length (bp) | 35,884 |
| Item | Number | Percent (%) |
|---|---|---|
| Complete BUSCO (C) | 3571 | 98.10 |
| Complete and single-copy BUSCO (S) | 3514 | 96.54 |
| Complete and duplicated BUSCO (D) | 57 | 1.57 |
| Fragmented BUSCO (F) | 21 | 0.58 |
| Missing BUSCO (M) | 48 | 1.32 |
| Total BUSCO groups searched | 3640 | 100.00 |
| Non-Code RNA Type | Copy Number | Total Length (bp) | Avg Length (bp) |
|---|---|---|---|
| tRNA | 2028 | 151,198 | 74.5552 |
| rRNA | 251 | 92,899 | 370.116 |
| snRNA | 473 | 61,713 | 130.471 |
| miRNA | 350 | 26,745 | 76.4143 |
| Type of Repeat | Length (bp) | % of Genome |
|---|---|---|
| SINEs | 11,580,806 | 0.68 |
| LINEs | 117,969,346 | 6.88 |
| L2/CR1/Rex | 77,550,358 | 4.53 |
| R1/LOA/Jockey | 7,296,609 | 0.43 |
| R2/R4/NeSL | 2,468,557 | 0.14 |
| RTE/Bov-B | 10,610,769 | 0.62 |
| L1/CIN4 | 13,198,944 | 0.77 |
| LTR | 37,360,206 | 2.18 |
| BEL/Pao | 2,804,533 | 0.16 |
| Ty1/Copia | 152,646 | 0.01 |
| Gypsy/DIRS1 | 13,611,972 | 0.79 |
| Retroviral | 6,496,637 | 0.38 |
| DNA transposons | 394,903,094 | 23.04 |
| hobo-Activator | 1,59,106,235 | 9.28 |
| Tc1-IS630-Pogo | 73,869,980 | 4.31 |
| PiggyBac | 6,409,425 | 0.37 |
| Tourist/Harbinger | 39,804,001 | 2.32 |
| Other | 3,827,180 | 0.22 |
| Rolling-circles | 10,770,335 | 0.63 |
| Unclassified | 280,705,119 | 16.38 |
| Satellite | 4,266,447 | 0.25 |
| Simple-repeat | 20,194,587 | 1.18 |
| Low complexity | 2,709,891 | 0.16 |
| Total | 880,459,831 | 51.38 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liang, Y.; Xian, L.; Pan, J.; Zhu, K.; Guo, H.; Liu, B.; Zhang, N.; Ou-Yang, Y.; Zhang, Q.; Zhang, D. De Novo Genome Assembly of the Whitespot Parrotfish (Scarus forsteni): A Valuable Scaridae Genomic Resource. Genes 2024, 15, 249. https://doi.org/10.3390/genes15020249
Liang Y, Xian L, Pan J, Zhu K, Guo H, Liu B, Zhang N, Ou-Yang Y, Zhang Q, Zhang D. De Novo Genome Assembly of the Whitespot Parrotfish (Scarus forsteni): A Valuable Scaridae Genomic Resource. Genes. 2024; 15(2):249. https://doi.org/10.3390/genes15020249
Chicago/Turabian StyleLiang, Yu, Lin Xian, Jinmin Pan, Kecheng Zhu, Huayang Guo, Baosuo Liu, Nan Zhang, Yan Ou-Yang, Qin Zhang, and Dianchang Zhang. 2024. "De Novo Genome Assembly of the Whitespot Parrotfish (Scarus forsteni): A Valuable Scaridae Genomic Resource" Genes 15, no. 2: 249. https://doi.org/10.3390/genes15020249