
Mitogenomic Characterization and Phylogenetic Placement of African Hind, Cephalopholis taeniops: Shedding Light on the Evolution of Groupers (Serranidae: Epinephelinae)

 , , , , , , , and
, , , , , , , and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Mitogenome Structure and Organization
2.2. Protein-Coding Genes
2.3. Substitutions Pattern and Codon Usage
2.4. Ribosomal RNA and Transfer RNA Genes
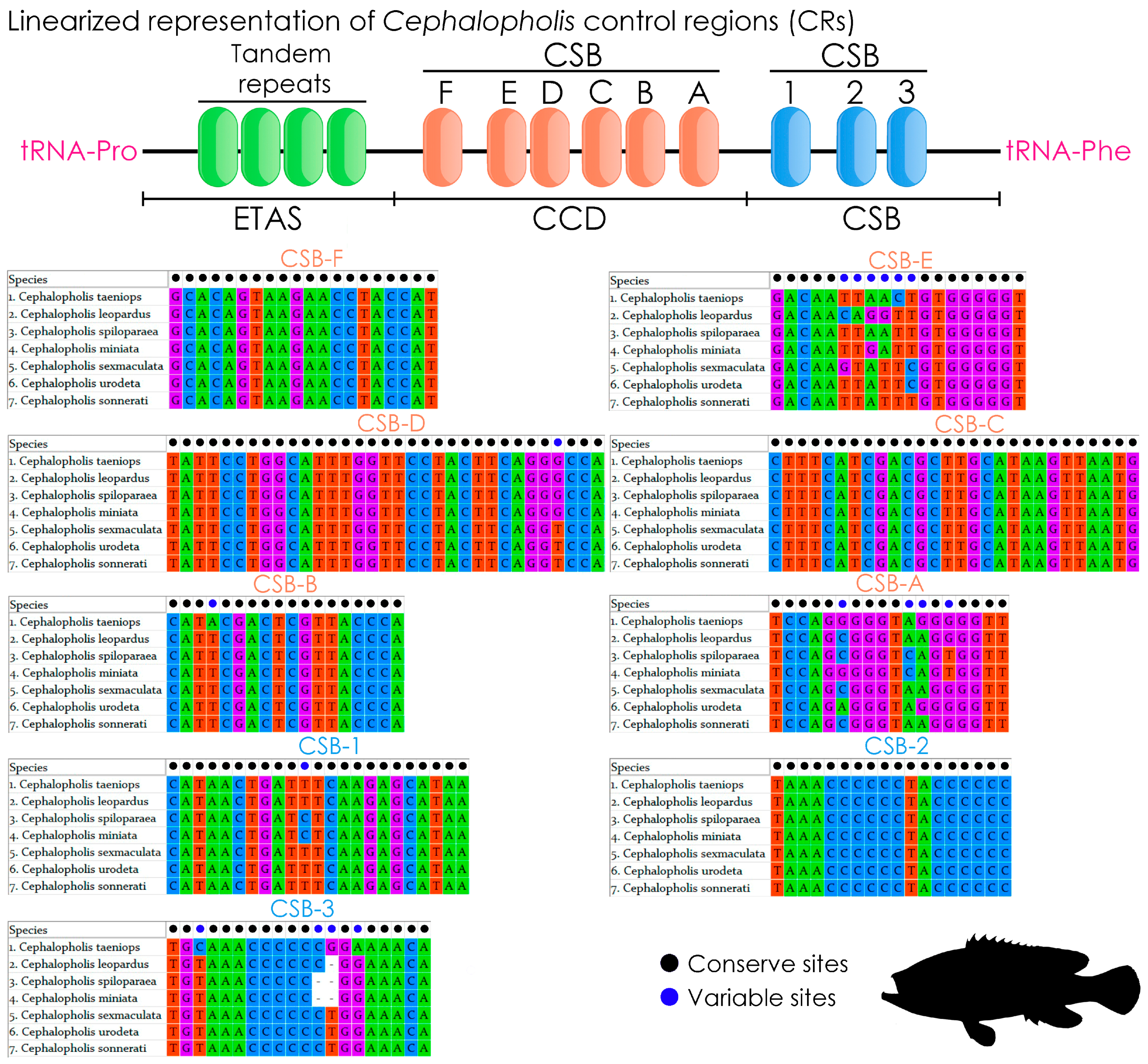
2.5. Features of Control Region
2.6. Genetic Distances and Phylogenetic Relationship
3. Materials and Methods
3.1. Sampling and Species Identification
3.2. DNA Extraction, Sequencing, and Assembly
3.3. Mitogenome Assembly and Validation of Control Region
3.4. Characterization and Comparative Analyses
3.5. Genetic Distance and Phylogenetic Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; de Bruijn, M.H.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef]
- Tzeng, C.S.; Hui, C.F.; Shen, S.C.; Huang, P.C. The complete nucleotide sequence of the Crossostoma lacustre mitochondrial genome: Conservation and variations among vertebrates. Nucleic Acids Res. 1992, 20, 4853–4858. [Google Scholar] [CrossRef]
- Harrison, R.G. Animal mitochondrial DNA as a genetic marker in population and evolutionary biology. Trends Ecol. Evol. 1989, 4, 6–11. [Google Scholar] [CrossRef]
- Miya, M.; Satoh, T.P.; Nishida, M. The phylogenetic position of toadfishes (order Batrachoidiformes) in the higher ray-finned fish as inferred from partitioned Bayesian analysis of 102 whole mitochondrial genome sequences. Biol. J. Linn. Soc. 2005, 85, 289–306. [Google Scholar] [CrossRef]
- Satoh, T.P.; Miya, M.; Mabuchi, K.; Nishida, M. Structure and variation of the mitochondrial genome of fishes. BMC Genom. 2016, 17, 719. [Google Scholar] [CrossRef] [PubMed]
- Heemstra, P.C.; Randall, J.E. FAO Species Catalogue. Groupers of the World (Family Serranidae, Subfamily Epinephelinae). An Annotated and Illustrated Catalogue of the Grouper, Rockcod, Hind, Coral Grouper and Lyretail Species Known to Date. FAO Fish. Synop. 1993, 125, 1–382. [Google Scholar]
- Fricke, R.; Eschmeyer, W.N.; Van der Laan, R. (Eds.) Eschmeyer’s Catalog of Fishes: Genera, Species. Available online: http://researcharchive.calacademy.org/research/ichthyology/catalog/fishcatmain.asp (accessed on 12 December 2023).
- Craig, M.T.; de Mitcheson, Y.S.; Heemstra, P.C. Groupers of the World: A Field and Market Guide; National Inquiry Services Centre: Grahamstown, South Africa, 2011; pp. 1–47. [Google Scholar]
- Vella, N.; Vella, A.; Darmanin, S.A. Morphological and genetic analyses of the first record of the Niger Hind, Cephalopholis nigri (Perciformes: Serranidae), in the Mediterranean Sea and of the African Hind, Cephalopholis taeniops, in Malta. Mar. Biodivers. Rec. 2016, 9, e99. [Google Scholar] [CrossRef]
- Ben Abdallah, A.; Ben Souissi, J.; Méjri, H.; Capapé, C.; Golani, D. First record of Cephalopholis taeniops (Valenciennes) in Mediterranean Sea. J. Fish Biol. 2007, 71, 610–614. [Google Scholar] [CrossRef]
- Salameh, P.; Sonin, O.; Golani, D. A first record of the African hind (Cephalopholis taeniops) (Pisces: Serranidae) in the Levant. Ann. Ser. Hist. Nat. 2009, 190, 151–154. [Google Scholar]
- Guidetti, P.; Giardina, F.; Azzurro, E. A new record of Cephalopholis taeniops in the Mediterranean Sea, with considerations on the Sicily channel as a biogeographical crossroad of exotic fish. Mar. Biodivers. Rec. 2010, 3, e13. [Google Scholar] [CrossRef]
- Brito, A.; Clemente, S.; Herrera, R. On the occurrence of the African hind, Cephalopholis taeniops, in the Canary Islands (eastern subtropical Atlantic): Introduction of large-sized demersal littoral fishes in ballast water of oil platforms? Biol. Invasions 2011, 13, 2185–2189. [Google Scholar] [CrossRef]
- Engin, S.; Irmak, E.; Seyhan, D. New record of the thermophilic Cephalopholis taeniops (Osteichthyes: Serranidae) in the Aegean Sea. Zool. Middle East. 2016, 62, 184–186. [Google Scholar] [CrossRef]
- Canes Garcia, L.; Rangel Moreira, C.; Carvalho Filho, A. First record of African Hind, Cephalopholis taeniops (Valenciennes, 1828) (Perciformes, Epinephelidae) in the South-western Atlantic. Check List 2018, 14, 961–965. [Google Scholar] [CrossRef]
- Luiz, O.J.; Woods, R.M.; Madin, E.M.; Madin, J.S. Predicting IUCN extinction risk categories for the world’s data deficient groupers (Teleostei: Epinephelidae). Conserv. Lett. 2016, 9, 342–350. [Google Scholar] [CrossRef]
- Burton, M.L.; Potts, J.C.; Carr, D.R. Age, growth and natural mortality of coney (Cephalopholis fulva) from the southeastern United States. PeerJ 2015, 3, e825. [Google Scholar] [CrossRef] [PubMed]
- Schemmel, E.M.; Donovan, M.K.; Wiggins, C.; Anzivino, M.; Friedlander, A.M. Reproductive life history of the introduced peacock grouper Cephalopholis argus in Hawaii. J. Fish Biol. 2016, 89, 1271–1284. [Google Scholar] [CrossRef] [PubMed]
- Gaither, M.R.; Bowen, B.W.; Bordenave, T.R.; Rocha, L.A.; Newman, S.J.; Gomez, J.A.; van Herwerden, L.; Craig, M.T. Phylogeography of the reef fish Cephalopholis argus (Epinephelidae) indicates Pleistocene isolation across the Indo-Pacific Barrier with contemporary overlap in The Coral Triangle. BMC Evol. Biol. 2011, 11, 189. [Google Scholar] [CrossRef]
- De Souza, A.S.; Dias Júnior, E.A.; Galetti, P.M., Jr.; Machado, E.G.; Pichorim, M.; Molina, W.F. Wide-range genetic connectivity of Coney, Cephalopholis fulva (Epinephelidae), through oceanic islands and continental Brazilian coast. An. Acad. Bras. Cienc. 2015, 87, 121–136. [Google Scholar] [CrossRef]
- Xie, Z.; Wang, D.; Jiang, S.; Peng, C.; Wang, Q.; Huang, C.; Li, S.; Lin, H.; Zhang, Y. Chromosome-Level Genome Assembly and Transcriptome Comparison Analysis of Cephalopholis sonnerati and Its Related Grouper Species. Biology 2022, 11, 1053. [Google Scholar] [CrossRef]
- Pondella, D.J., 2nd; Craig, M.T.; Franck, J.P. The phylogeny of Paralabrax (Perciformes: Serranidae) and allied taxa inferred from partial 16S and 12S mitochondrial ribosomal DNA sequences. Mol. Phylogenet. Evol. 2003, 29, 176–184. [Google Scholar] [CrossRef]
- Craig, M.T.; Hastings, P.A. A molecular phylogeny of the groupers of the subfamily Epinephelinae (Serranidae) with a revised classification of the Epinephelini. Ichthyol. Res. 2007, 54, 1–17. [Google Scholar] [CrossRef]
- Gill, A.C.; Pogonoski, J.J.; Johnson, J.W.; Tea, Y.K. Three new species of Australian anthiadine fishes, with comments on the monophyly of Pseudanthias Bleeker (Teleostei: Serranidae). Zootaxa 2021, 4996, 49–82. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.T.; Viviani, J. Pseudogramma polyacantha complex (Serranidae, tribe Grammistini): DNA barcoding results lead to the discovery of three cryptic species, including two new species from French Polynesia. Zootaxa 2016, 4111, 246–260. [Google Scholar]
- Harrison, H.B.; Feldheim, K.A.; Jones, G.P.; Ma, K.; Mansour, H.; Perumal, S.; Williamson, D.H.; Berumen, M.L. Validation of microsatellite multiplexes for parentage analysis and species discrimination in two hybridizing species of coral reef fish (Plectropomus spp., Serranidae). Ecol. Evol. 2014, 4, 2046–2057. [Google Scholar] [CrossRef]
- Craig, M.T.; Pondella, D.J.; Franck, J.P.; Hafner, J.C. On the status of the Serranid fish genus Epinephelus: Evidence for paraphyly based upon 16S rDNA sequence. Mol. Phylogenet. Evol. 2001, 19, 121–130. [Google Scholar] [CrossRef]
- Ding, S.; Zhuang, X.; Guo, F.; Wang, J.; Su, Y.; Zhang, Q.; Li, Q. Molecular phylogenetic relationships of China Seas groupers based on cytochrome b gene fragment sequences. Sci. China C Life Sci. 2006, 49, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.Y.; Craig, M.T. An Inconvenient Monophyly: An Update on the Taxonomy of the Groupers (Epinephelidae). Copeia 2018, 106, 443–456. [Google Scholar] [CrossRef]
- Stepien, C.A.; Rosenblatt, R.H.; Bargmeyer, B.A. Phylogeography of the spotted sand bass, Paralabrax maculatofasciatus: Divergence of Gulf of California and Pacific Coast populations. Evolution 2001, 55, 1852–1862. [Google Scholar]
- Liu, J.X.; Gao, T.X.; Yokogawa, K.; Zhang, Y.P. Differential population structuring and demographic history of two closely related fish species, Japanese sea bass (Lateolabrax japonicus) and spotted sea bass (Lateolabrax maculatus) in Northwestern Pacific. Mol. Phylogenet. Evol. 2006, 39, 799–811. [Google Scholar] [CrossRef]
- Gauthier, D.T.; Audemard, C.A.; Carlsson, J.E.; Darden, T.L.; Denson, M.R.; Reece, K.S.; Carlsson, J. Genetic population structure of US Atlantic coastal striped bass (Morone saxatilis). J. Hered. 2013, 104, 510–520. [Google Scholar] [CrossRef]
- Buchholz-Sørensen, M.; Vella, A. Population Structure, Genetic Diversity, Effective Population Size, Demographic History and Regional Connectivity Patterns of the Endangered Dusky Grouper, Epinephelus marginatus (Teleostei: Serranidae), within Malta’s Fisheries Management Zone. PLoS ONE 2016, 11, e0159864. [Google Scholar] [CrossRef] [PubMed]
- Puebla, O.; Bermingham, E.; Guichard, F. Population genetic analyses of Hypoplectrus coral reef fishes provide evidence that local processes are operating during the early stages of marine adaptive radiations. Mol. Ecol. 2008, 17, 1405–1415. [Google Scholar] [CrossRef] [PubMed]
- Bariche, M.; Torres, M.; Smith, C.; Sayar, N.; Azzurro, E.; Baker, R.; Bernardi, G. Red Sea fishes in the Mediterranean Sea: A preliminary investigation of a biological invasion using DNA barcoding. J. Biogeogr. 2015, 42, 2363–2373. [Google Scholar] [CrossRef]
- Galal-Khallaf, A.; Osman, A.G.M.; El-Ganainy, A.; Farrag, M.M.; Mohammed-AbdAllah, E.; Moustafa, M.A.; Mohammed-Geba, K. Mitochondrial genetic markers for authentication of major Red Sea grouper species (Perciformes: Serranidae) in Egypt: A tool for enhancing fisheries management and species conservation. Gene 2019, 689, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Duranton, M.; Allal, F.; Fraïsse, C.; Bierne, N.; Bonhomme, F.; Gagnaire, P.A. The origin and remolding of genomic islands of differentiation in the European sea bass. Nat. Commun. 2018, 9, 2518. [Google Scholar] [CrossRef]
- Sun, C.F.; Zhang, X.H.; Dong, J.J.; You, X.X.; Tian, Y.Y.; Gao, F.Y.; Zhang, H.T.; Shi, Q.; Ye, X. Whole-genome resequencing reveals recent signatures of selection in five populations of largemouth bass (Micropterussalmoides). Zool. Res. 2023, 44, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Coulmance, F.; Akkaynak, D.; Le Poul, Y.; Höppner, M.P.; McMillan, W.O.; Puebla, O. Phenotypic and genomic dissection of colour pattern variation in a reef fish radiation. Mol. Ecol. 2023. [Google Scholar] [CrossRef]
- Zhuang, X.; Qu, M.; Zhang, X.; Ding, S. A comprehensive description and evolutionary analysis of 22 Grouper (Perciformes, Epinephelidae) mitochondrial genomes with emphasis on two novel genome organizations. PLoS ONE 2013, 8, e73561. [Google Scholar] [CrossRef]
- Li, J.L.; Liu, M.; Wang, Y.Y. Complete mitochondrial genome of the chocolate hind Cephalopholis boenak (Pisces: Perciformes). Mitochondrial DNA 2014, 25, 167–168. [Google Scholar] [CrossRef]
- Hsiao, S.T.; Chen, K.S.; Tseng, C.T.; Wu, C.L. Complete mitochondrial genome of the sixblotch hind Cephalopholis sexmaculata (Pisces: Perciformes). Mitochondrial DNA Part A DNA Mapp. Seq. Anal. 2016, 27, 1018–1019. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Huang, H.; Gao, Y. Complete mitochondrial genome of darkfin hind Cephalopholis urodeta (Perciformes, Epinephelidae). Mitochondrial DNA B Resour. 2016, 1, 913–916. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Zhang, Y.; Gong, L.; Gao, Y. Characterization of the complete mitochondrial genome of Cephalopholis miniata (Perciformes, Serranidae) and its phylogenetic analysis. Mitochondrial DNA B Resour. 2021, 6, 1976–1978. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Ye, P.; Liu, M.; Zhang, Y.; Feng, H.; Liu, J.; Zhou, H.; Wang, J.; Chen, X. Comparative Analysis of Four Complete Mitochondrial Genomes of Epinephelidae (Perciformes). Genes 2022, 13, 660. [Google Scholar] [CrossRef] [PubMed]
- Miya, M.; Kawaguchi, A.; Nishida, M. Mitogenomic exploration of higher teleostean phylogenies: A case study for moderate-scale evolutionary genomics with 38 newly determined complete mitochondrial DNA sequences. Mol. Biol. Evol. 2001, 18, 1993–2009. [Google Scholar] [CrossRef]
- Saitoh, K.; Sado, T.; Mayden, R.L.; Hanzawa, N.; Nakamura, K.; Nishida, M.; Miya, M. Mitogenomic evolution and interrelationships of the Cypriniformes (Actinopterygii: Ostariophysi): The first evidence toward resolution of higher-level relationships of the world’s largest freshwater fish clade based on 59 whole mitogenome sequences. J. Mol. Evol. 2006, 63, 826–841. [Google Scholar] [CrossRef]
- Yamanoue, Y.; Miya, M.; Matsuura, K.; Yagishita, N.; Mabuchi, K.; Sakai, H.; Katoh, M.; Nishida, M. Phylogenetic position of tetraodontiform fishes within the higher teleosts: Bayesian inferences based on 44 whole mitochondrial genome sequences. Mol. Phylogenet. Evol. 2007, 45, 89–101. [Google Scholar] [CrossRef]
- Saccone, C.; Pesole, G.; Sbisá, E. The main regulatory region of mammalian mitochondrial DNA: Structure-function model and evolutionary pattern. J. Mol. Evol. 1991, 33, 83–91. [Google Scholar] [CrossRef]
- Bernt, M.; Braband, A.; Schierwater, B.; Stadler, P.F. Genetic aspects of mitochondrial genome evolution. Mol. Phylogenet. Evol. 2013, 69, 328–338. [Google Scholar] [CrossRef]
- Sato, Y.; Miya, M.; Fukunaga, T.; Sado, T.; Iwasaki, W. MitoFish and MiFish Pipeline: A Mitochondrial Genome Database of Fish with an Analysis Pipeline for Environmental DNA Metabarcoding. Mol. Biol. Evol. 2018, 35, 1553–1555. [Google Scholar] [CrossRef]
- Bowen, B.W.; Rocha, L.A.; Toonen, R.J.; Karl, S.A.; ToBo Laboratory. The origins of tropical marine biodiversity. Trends Ecol. Evol. 2013, 28, 359–366. [Google Scholar] [CrossRef]
- Myers, N.; Mittermeier, R.A.; Mittermeier, C.G.; da Fonseca, G.A.; Kent, J. Biodiversity hotspots for conservation priorities. Nature 2000, 403, 853–858. [Google Scholar] [CrossRef]
- Miller, E.C.; Hayashi, K.T.; Song, D.; Wiens, J.J. Explaining the ocean’s richest biodiversity hotspot and global patterns of fish diversity. Proc. Biol. Sci. 2018, 285, 20181314. [Google Scholar] [CrossRef]
- Fan, H.; Huang, M.; Chen, Y.; Zhou, W.; Hu, Y.; Wei, F. Conservation priorities for global marine biodiversity across multiple dimensions. Natl. Sci. Rev. 2022, 10, nwac241. [Google Scholar] [CrossRef]
- Simard, F.; Laffoley, D.; Baxter, J.M. (Eds.) Marine Protected Areas and Climate Change: Adaptation and Mitigation Synergies, Opportunities and Challenges; IUCN: Gland, Switzerland, 2016; 52p. [Google Scholar]
- Wang, X.; Wang, Y.; Zhang, Y.; Yu, H.; Tong, J. Evolutionary analysis of cyprinid mitochondrial genomes: Remarkable variation and strong adaptive evolution. Front. Genet. 2016, 7, 156. [Google Scholar]
- Kosiol, C.; Vinar, T.; da Fonseca, R.R.; Hubisz, M.J.; Bustamante, C.D.; Nielsen, R.; Siepel, A. Patterns of positive selection in six Mammalian genomes. PLoS Genet. 2008, 4, e1000144. [Google Scholar] [CrossRef] [PubMed]
- Foote, A.D.; Morin, P.A.; Durban, J.W.; Pitman, R.L.; Wade, P.; Willerslev, E.; Gilbert, M.T.; da Fonseca, R.R. Positive selection on the killer whale mitogenome. Biol. Lett. 2011, 7, 116–118. [Google Scholar] [CrossRef] [PubMed]
- Hirsh, A.E.; Fraser, H.B. Protein dispensability and rate of evolution. Nature 2001, 411, 1046–1049. [Google Scholar] [CrossRef] [PubMed]
- Bloom, J.D.; Labthavikul, S.T.; Otey, C.R.; Arnold, F.H. Protein stability promotes evolvability. Proc. Natl. Acad. Sci. USA 2006, 103, 5869–5874. [Google Scholar] [CrossRef] [PubMed]
- Montoya-Burgos, J.I. Patterns of Positive Selection and Neutral Evolution in the Protein-Coding Genes of Tetraodon and Takifugu. PLoS ONE 2011, 6, e24800. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Bielawski, J.P. Statistical methods for detecting molecular adaptation. Trends Ecol. Evol. 2000, 15, 496–503. [Google Scholar] [CrossRef]
- Yang, Z.H.; Nielsen, R. Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol. Biol. Evol. 2000, 17, 32–43. [Google Scholar] [CrossRef]
- Zhu, K.C.; Liang, Y.Y.; Wu, N.; Guo, H.Y.; Zhang, N.; Jiang, S.G.; Zhang, D.C. Sequencing and characterization of the complete mitochondrial genome of Japanese Swellshark (Cephalloscyllium umbratile). Sci. Rep. 2017, 7, 15299. [Google Scholar] [CrossRef]
- Nei, M.; Kumar, S. Molecular Evolution and Phylogenetics; Oxford University Press: New York, NY, USA, 2000. [Google Scholar]
- Sato, N.S.; Hirabayashi, N.; Agmon, I.; Yonath, A.; Suzuki, T. Comprehensive genetic selection revealed essential bases in the peptidyl-transferase center. Proc. Natl. Acad. Sci. USA 2006, 103, 15386–15391. [Google Scholar] [CrossRef]
- Ojala, D.; Montoya, J.; Attardi, G. tRNA punctuation model of RNA processing in human mitochondria. Nature 1981, 290, 470–474. [Google Scholar] [CrossRef]
- Varani, G.; McClain, W.H. The G-U wobble base pair: A fundamental building block of RNA structure crucial to RNA function in diverse biological systems. EMBO Rep. 2000, 1, 18–23. [Google Scholar] [CrossRef]
- Cantatore, P.; Gadaleta, M.N.; Roberti, M.; Saccone, C.; Wilson, A.C. Duplication and remoulding of tRNA genes during the evolutionary rearrangement of mitochondrial genomes. Nature 1987, 329, 853–855. [Google Scholar] [CrossRef] [PubMed]
- Ponce, M.; Infante, C.; Jiménez-Cantizano, R.M.; Pérez, L.; Manchado, M. Complete mitochondrial genome of the blackspot seabream, Pagellus bogaraveo (Perciformes: Sparidae), with high levels of length heteroplasmy in the WANCY region. Gene 2008, 409, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.J.; Conroy, J.; Howell, W.H.; Kocher, T.D. Structure and evolution of teleost mitochondrial control regions. J. Mol. Evol. 1995, 41, 54–66. [Google Scholar] [CrossRef] [PubMed]
- San Mauro, D.; Gower, D.J.; Zardoya, R.; Wilkinson, M. A hotspot of gene order rearrangement by tandem duplication and random loss in the vertebrate mitochondrial genome. Mol. Biol. Evol. 2006, 23, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Springer, M.S.; Douzery, E. Secondary structure and patterns of evolution among mammalian mitochondrial 12S rRNA molecules. J. Mol. Evol. 1996, 43, 357–373. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.Y.; Lee, S.C. Secondary structure of mitochondrial 12S rRNA among fish and its phylogenetic applications. Mol. Biol. Evol. 2002, 19, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, X.; Kong, X.; Zhao, K.; He, S.; Mayden, R.L. Variation patterns of the mitochondrial 16S rRNA gene with secondary structure constraints and their application to phylogeny of cyprinine fishes (Teleostei: Cypriniformes). Mol. Phylogenet. Evol. 2008, 47, 472–487. [Google Scholar] [CrossRef]
- Kundu, S.; De Alwis, P.S.; Kim, A.R.; Lee, S.R.; Kang, H.-E.; Go, Y.; Gietbong, F.Z.; Wibowo, A.; Kim, H.-W. Mitogenomic Characterization of Cameroonian Endemic Coptodon camerunensis (Cichliformes: Cichlidae) and Matrilineal Phylogeny of Old-World Cichlids. Genes 2023, 14, 1591. [Google Scholar] [CrossRef]
- Kundu, S.; Palimirmo, F.S.; Kang, H.-E.; Kim, A.R.; Lee, S.R.; Gietbong, F.Z.; Song, S.H.; Kim, H.-W. Insights into the Mitochondrial Genetic Makeup and Miocene Colonization of Primitive Flatfishes (Pleuronectiformes: Psettodidae) in the East Atlantic and Indo-West Pacific Ocean. Biology 2023, 12, 1317. [Google Scholar] [CrossRef]
- Tine, M.; Kuhl, H.; Gagnaire, P.A.; Louro, B.; Desmarais, E.; Martins, R.S.; Hecht, J.; Knaust, F.; Belkhir, K.; Klages, S.; et al. European Sea bass genome and its variation provide insights into adaptation to euryhalinity and speciation. Nat. Commun. 2014, 5, 5770. [Google Scholar] [CrossRef]
- Cheung, W.W.L.; Frölicher, T.L.; Lam, V.W.Y.; Oyinlola, M.A.; Reygondeau, G.; Sumaila, U.R.; Tai, T.C.; The, L.C.L.; Wabnitz, C.C.C. Marine high temperature extremes amplify the impacts of climate change on fish and fisheries. Sci. Adv. 2021, 7, eabh0895. [Google Scholar] [CrossRef]
- Waterhouse, L.; Heppell, S.A.; Pattengill-Semmens, C.V.; McCoy, C.; Bush, P.; Johnson, B.C.; Semmens, B.X. Recovery of critically endangered Nassau grouper (Epinephelus striatus) in the Cayman Islands following targeted conservation actions. Proc. Natl. Acad. Sci. USA 2020, 117, 1587–1595. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo Metazoan Mitochondrial Genome Annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Iwasaki, W.; Fukunaga, T.; Isagozawa, R.; Yamada, K.; Maeda, Y.; Satoh, T.P.; Sado, T.; Mabuchi, K.; Takeshima, H.; Miya, M.; et al. MitoFish and MitoAnnotator: A mitochondrial genome database of fish with an accurate and automatic annotation pipeline. Mol. Biol. Evol. 2013, 30, 2531–2540. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–359. [Google Scholar] [CrossRef]
- Xia, X. DAMBE6: New tools for microbial genomics, phylogenetics and molecular evolution. J. Hered. 2017, 108, 431–437. [Google Scholar] [CrossRef]
- Laslett, D.; Canbäck, B. ARWEN, a program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 2008, 24, 172–175. [Google Scholar] [CrossRef]
- Chan, P.P.; Lin, B.Y.; Mak, A.J.; Lowe, T.M. tRNAscan-SE 2.0: Improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 2021, 49, 9077–9096. [Google Scholar] [CrossRef]
- Thompson, J.D.; Gibson, T.J.; Plewniak, F.; Jeanmougin, F.; Higgins, D.G. The CLUSTAL_X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 25, 4876–4882. [Google Scholar] [CrossRef]
- Shen, M.; Shi, X.; Qu, M.; Chen, J. Complete mitochondrial genome of squaretail coralgrouper Plectropomus areolatus (Perciformes, Epinephelidae). Mitochondrial DNA 2013, 24, 365–367. [Google Scholar] [CrossRef]
- Wang, H.; Guo, L.; Ding, S.X. The complete mitochondrial genome of Diploprion bifasciatum (Perciformes, Serranidae). Mitochondrial DNA Part A DNA Mapp. Seq. Anal. 2016, 27, 3137–3138. [Google Scholar] [CrossRef]
- Hoban, M.L.; Whitney, J.; Collins, A.G.; Meyer, C.; Murphy, K.R.; Reft, A.J.; Bemis, K.E. Skimming for barcodes: Rapid production of mitochondrial genome and nuclear ribosomal repeat reference markers through shallow shotgun sequencing. PeerJ 2022, 10, e13790. [Google Scholar] [CrossRef]
- Vella, A.; Vella, N.; Acosta-Díaz, C. The first complete mitochondrial genomes for Serranus papilionaceus and Serranus scriba, and their phylogenetic position within Serranidae. Mol. Biol. Rep. 2022, 49, 6295–6302. [Google Scholar] [CrossRef]
- Vences, M.; Miralles, A.; Brouillet, S.; Ducasse, J.; Fedosov, A.; Kharchev, V.; Kostadinov, I.; Kumari, S.; Patmanidis, S.; Scherz, M.D.; et al. iTaxoTools 0.1: Kickstarting a specimen-based software toolkit for taxonomists. Megataxa 2021, 6, 77–92. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. JModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef]
- Miller, M.A.; Schwartz, T.; Pickett, B.E.; He, S.; Klem, E.B.; Scheuermann, R.H.; Passarotti, M.; Kaufman, S.; O’Leary, M.A. A RESTful API for Access to Phylogenetic Tools via the CIPRES Science Gateway. Evol. Bioinform. 2015, 11, 43–48. [Google Scholar] [CrossRef]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New Methods for Selecting Partitioned Models of Evolution for Molecular and Morphological Phylogenetic Analyses. Mol. Biol. Evol. 2016, 34, 772–773. [Google Scholar] [CrossRef]
- Trifinopoulos, J.; Nguyen, L.-T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL): An online tool for phylogenetic tree display and annotation. Bioinformatics 2007, 23, 127–128. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Start | Stop | Strand | Size (bp) | Intergenic Nucleotide | Anticodon | Start Codon | Stop Codon |
|---|---|---|---|---|---|---|---|---|
| tRNA-Phe (F) | 1 | 70 | H | 70 | 0 | TTC | . | . |
| 12S rRNA | 71 | 1023 | H | 953 | 0 | . | . | . |
| tRNA-Val (V) | 1024 | 1095 | H | 72 | 0 | GTA | . | . |
| 16S rRNA | 1096 | 2807 | H | 1712 | 0 | . | . | . |
| tRNA-Leu (L2) | 2808 | 2882 | H | 75 | 0 | TTA | . | . |
| ND1 | 2883 | 3857 | H | 975 | 5 | . | ATG | TAA |
| tRNA-Ile (I) | 3863 | 3932 | H | 70 | −1 | ATC | . | . |
| tRNA-Gln (Q) | 3932 | 4002 | L | 71 | 0 | CAA | . | . |
| tRNA-Met (M) | 4003 | 4072 | H | 70 | 0 | ATG | . | . |
| ND2 | 4073 | 5118 | H | 1046 | 0 | . | ATG | TA- |
| tRNA-Trp (W) | 5119 | 5189 | H | 71 | 1 | TGA | . | . |
| tRNA-Ala (A) | 5191 | 5259 | L | 69 | 0 | GCA | . | . |
| tRNA-Asn (N) | 5260 | 5332 | L | 73 | 37 | AAC | . | . |
| tRNA-Cys (C) | 5370 | 5437 | L | 68 | 0 | TGC | . | . |
| tRNA-Tyr (Y) | 5438 | 5508 | L | 71 | 1 | TAC | . | . |
| COI | 5510 | 7060 | H | 1551 | 0 | . | GTG | TAA |
| tRNA-Ser (S2) | 7061 | 7131 | L | 71 | 1 | TCA | . | . |
| tRNA-Asp (D) | 7133 | 7205 | H | 73 | 8 | GAC | . | . |
| COII | 7214 | 7904 | H | 691 | 0 | . | ATG | T-- |
| tRNA-Lys (K) | 7905 | 7977 | H | 73 | 1 | AAA | . | . |
| ATP8 | 7979 | 8146 | H | 168 | −10 | . | ATG | TAA |
| ATP6 | 8137 | 8819 | H | 683 | 0 | . | TTG | TA- |
| COIII | 8820 | 9604 | H | 785 | 0 | . | ATG | TA- |
| tRNA-Gly (G) | 9605 | 9676 | H | 72 | 0 | GGA | . | . |
| ND3 | 9677 | 10,025 | H | 349 | 0 | . | ATG | T-- |
| tRNA-Arg (R) | 10,026 | 10,094 | H | 69 | 0 | CGA | . | . |
| ND4L | 10,095 | 10,391 | H | 297 | −7 | . | ATG | TAA |
| ND4 | 10,385 | 11,765 | H | 1381 | 0 | . | ATG | T-- |
| tRNA-His (H) | 11,766 | 11,835 | H | 70 | 0 | CAC | . | . |
| tRNA-Ser (S1) | 11,836 | 11,907 | H | 72 | 6 | AGC | . | . |
| tRNA-Leu (L1) | 11,914 | 11,986 | H | 73 | 0 | CTA | . | . |
| ND5 | 11,987 | 13,825 | H | 1839 | −4 | . | ATG | TAA |
| ND6 | 13,822 | 14,343 | L | 522 | 0 | . | ATG | TAA |
| tRNA-Glu (E) | 14,344 | 14,412 | L | 69 | 4 | GAA | . | . |
| Cyt b | 14,417 | 15,557 | H | 1141 | 0 | . | ATG | T-- |
| tRNA-Thr (T) | 15,558 | 15,630 | H | 73 | −1 | ACA | . | . |
| tRNA-Pro (P) | 15,630 | 15,699 | L | 70 | 0 | CCA | . | . |
| Control region (CR) | 15,700 | 16,572 | H | 873 | . | . | . | . |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kundu, S.; Kang, H.-E.; Kim, A.R.; Lee, S.R.; Kim, E.-B.; Amin, M.H.F.; Andriyono, S.; Kim, H.-W.; Kang, K. Mitogenomic Characterization and Phylogenetic Placement of African Hind, Cephalopholis taeniops: Shedding Light on the Evolution of Groupers (Serranidae: Epinephelinae). Int. J. Mol. Sci. 2024, 25, 1822. https://doi.org/10.3390/ijms25031822
Kundu S, Kang H-E, Kim AR, Lee SR, Kim E-B, Amin MHF, Andriyono S, Kim H-W, Kang K. Mitogenomic Characterization and Phylogenetic Placement of African Hind, Cephalopholis taeniops: Shedding Light on the Evolution of Groupers (Serranidae: Epinephelinae). International Journal of Molecular Sciences. 2024; 25(3):1822. https://doi.org/10.3390/ijms25031822
Chicago/Turabian StyleKundu, Shantanu, Hye-Eun Kang, Ah Ran Kim, Soo Rin Lee, Eun-Bi Kim, Muhammad Hilman Fu’adil Amin, Sapto Andriyono, Hyun-Woo Kim, and Kyoungmi Kang. 2024. "Mitogenomic Characterization and Phylogenetic Placement of African Hind, Cephalopholis taeniops: Shedding Light on the Evolution of Groupers (Serranidae: Epinephelinae)" International Journal of Molecular Sciences 25, no. 3: 1822. https://doi.org/10.3390/ijms25031822