 Silvia Turco1*†
Silvia Turco1*† Alessandro Grottoli2†
Alessandro Grottoli2† Mounira Inas Drais1
Mounira Inas Drais1 Carlo De Spirito1
Carlo De Spirito1 Luigi Faino3
Luigi Faino3 Massimo Reverberi3
Massimo Reverberi3 Valerio Cristofori1
Valerio Cristofori1 Angelo Mazzaglia1*
Angelo Mazzaglia1*
- 1Dipartimento di Scienze Agrarie e Forestali, Università degli Studi della Tuscia, Viterbo, Italy
- 2Consiglio per la Ricerca in Agricoltura e l’Analisi dell’Economia Agraria, Centro di Ricerca Difesa e Certificazione (CREA-DC), Rome, Italy
- 3Dipartimento di Biologia Ambientale, Sapienza Università di Roma, Rome, Italy
In summer 2019, during a survey on the health status of a hazelnut orchard located in the Tuscia area (the province of Viterbo, Latium, Italy), nuts showing symptoms, such as brown-grayish spots at the bottom of the nuts progressing upward to the apex, and necrotic patches on the bracts and, sometimes, on the petioles, were found and collected for further studies. This syndrome is associated with the nut gray necrosis (NGN), whose main causal agent is Fusarium lateritium. Aiming to increase knowledge about this fungal pathogen, the whole-genome sequencing of a strain isolated from symptomatic hazelnut was performed using long Nanopore reads technology in combination with the higher precision of the Illumina reads, generating a high-quality genome assembly. The following phylogenetic and comparative genomics analysis suggested that this isolate is caused by the F. tricinctum species complex rather than F. lateritium one, as initially hypothesized. Thus, this study demonstrates that different Fusarium species can infect Corylus avellana producing the same symptomatology. In addition, it sheds light onto the genetic features of the pathogen in subject, clarifying facets about its biology, epidemiology, infection mechanisms, and host spectrum, with the future objective to develop specific and efficient control strategies.
Introduction
Corylus avellana L. (hazelnut) is a shrub species belonging to the Betulaceae family. Italy is the second largest hazelnut producer in the world, with an average production of about 140,000 t/year spread among four regions (Campania, Latium, Sicily, and Piedmont), behind Turkey (ISTAT1, FAOstat Agriculture Data2). In the last decades, a fruit rot causing considerable yield losses has been observed and described as a new disease. The symptomatic fruits were characterized by the presence of brown-grayish spots at the bottom of the nuts progressing upward to the apex, and necrotic patches on the bracts and, less often, on the petioles (Belisario and Santori, 2009). Based on these symptoms, the disease has been named nut gray necrosis (NGN) and associated with Fusarium lateritium Nees [Gibberella baccata (Wallr.) Sacc.] as its causal agent (Santori et al., 2010; Vitale et al., 2011). Fusarium is a large cosmopolitan genus of filamentous ascomycetes fungi, ranked as one of the most economically destructive and species-rich groups in the world, including plant pathogens, saprophytes, and endophytes species, among others (O’Donnell et al., 2013, 2015). Among the numerous species, F. lateritium has been reported on numerous hosts, including woody fruit trees as well as shrubs and herbaceous plants, where it could induce wilting, tip or branch dieback, and cankers. F. lateritium has also been reported as the causal agent of twig canker on hazelnut, and fruit rot on walnut (Wollenweber, 1931) and olive (Elia, 1964). Several pathogenicity tests were conducted, supporting the involvement of this fungus in the NGN disease and twig canker of hazelnut (Belisario and Santori, 2009).
In late summer 2019, a survey on the health status of a hazelnut orchard located in the Tuscia area (the province of Viterbo, Latium, Italy) was carried out in order to combine an agronomic evaluation of the state of the field approach with a Supervisory Control and Data Acquisition (SCADA) system for the precision farming of orchards. Nuts showing NGN symptoms were found and collected for further laboratory analysis and molecular characterization.
Aiming to increase knowledge about this fungal pathogen, the genome of one fungal strain isolated from a typical NGN diseased nut was sequenced using both long- and short-reads sequencing technologies. The resulting genome when compared with other available Fusarium genomes showed that, despite a morphological similarity with F. lateritium, this isolate is related to the F. tricinctum species complex rather than to the F. lateritium one. Thus, this study aims to provide new insights about the complexity of Fusarium species that infect tissues and fruits of hazelnut and to better understand the genetics behind the pathogenic mechanisms of this fungal strain. The new information achieved represents the basis for a better focused and effective control strategy of this disease.
Materials and Methods
Fungal Isolation
Symptomatic nuts (Tonda Gentile Romana cv) were collected from a mature hazelnut orchard located in the Viterbo area (VT) (Latium, Italy; latitude 42°16′00.0″, longitude 12°17′00.0″, altitude 275 m). Small fragments of approximately 2 mm2 were taken from the nut surface and placed onto potato dextrose agar (PDA) plates supplemented with 0.2 g⋅L–1 streptomycin sulfate. The Petri plates were then incubated at 25°C until the fungal colonies were grown enough to be singularly further transferred onto new PDA plates to finally obtain monosporal isolates.
Morphological and Molecular Identification
For morphological characterization, the cultures were grown on Spezieller Nahrstoffarmer agar (SNA) (Leslie and Summerell, 2006). After 10 days, the cultures were examined using a Nikon SMZ128 stereomicroscope (Tokyo, Japan), and the images were captured using Alexasoft TPS5000H CMOS camera (Florence, Italy). For microscopic analysis, the samples were prepared by mixing with lactophenol blue dye using Leica DM6 B optical microscope (Wetzlar, Germany), the images were captured using Leica DFC 7000 T camera (Wetzlar, Germany), elaborated using Leica Application Suite X program (version 4.12) (Wetzlar, Germany), and all the morphological characteristics were evaluated according to The Fusarium Laboratory Manual (Leslie and Summerell, 2006).
For each isolate, about 100 mg of mycelium was put into a sterile 2 ml microtube with 1 ml of lysis buffer (1 mM EDTA pH 8, 10 mM Tris–HCl pH 8, 100 mM NaCl, SDS 1%) for DNA extraction. After 5 min of centrifugation at 12,000 rpm, the supernatant was transferred into a new microtube, and the DNA was precipitated with isopropanol and 70% ethanol. To characterize the isolates at the genus/species level, two genes were amplified using two different pairs of primers, one targeting the internal transcribed spacer (ITS) (White et al., 1990) and the second one targeting the Fusarium specific translation elongation factor 1-alpha (Edel-Hermann et al., 2015), and the amplicons were Sanger sequenced at Eurofins genomics (Eurofins Genomics GmbH, Konstanz, Germany).
Pathogenicity Tests
To confirm that the isolate in subject was responsible for the observed symptoms on fruits, the following pathogenicity tests were carried out. For inoculum preparation, the PT isolate of the Fusarium sp. was grown on SNA at 25°C for 10 days (Leslie and Summerell, 2006). Then, conidia were scraped from the fungal cultures and filtered through layers of cheesecloth to remove mycelial fragments. The resulting conidial suspension was quantified using a hemocytometer and diluted to a final concentration of 106 spore/ml. Inoculations were performed using two techniques: (i) inoculation of young to fully formed fruits with 20 μl of conidial suspension (106 conidia/ml) dropped between nut and bracts; and (ii) inoculation of the hazelnut mesocarp with 20 μl of conidial suspension (106 conidia/ml). Control fruits were inoculated with water. Inoculated and control fruits were placed in a moist chamber at room temperature for 12 days. The appearance of any symptoms was monitored daily.
High Molecular Weight DNA Extraction and Genome Sequencing
The fungal culture was initiated using ∼1 × 106 conidia inoculated in 100 ml of Czapek Dox Yeast broth (Leslie and Summerell, 2006) and grown for 3 days at 25°C. Next, the genomic DNA was extracted following a modified Cetrimioum bromide (CTAB) method (Graham and Henry, 1997) and performed as described below.
The total mycelium grown in liquid media was lyophilized using a vacuum pump. An aliquot of 250 mg of lyophilized mycelium was fine grounded using a pestle and mortar in liquid nitrogen and transferred into a 2 ml tube. Then, 500 μl of CTAB buffer (10% CTAB, 25 mM EDTA pH 8.0, 200 mM Tris–HCl pH 8.0, 2.50 M NaCl) and 5 μl proteinase K (20 mg/ml) were added to the sample and the solution was gently mixed and incubated overnight at 56°C. After 5 min of agitated incubation with 500 μl of phenol:chloroform:isoamyl alcohol (25:24:1) and 10 min of 12,000 rpm centrifugation, the aqueous clear phase was transferred to a new tube and incubated with 500 μl of chloroform:isoamyl alcohol (24:1). After 10 min of centrifugation at 12,000 rpm, the aqueous phase was collected into a new tube and 0.6 VOL of cold isopropanol was added, followed by 30 min of centrifugation at 12,000 rpm for the DNA precipitation. Two cleaning/precipitation steps using 1 ml of 70% cold ethanol were performed by 10 min of centrifugation at 12,000 rpm. The pellet was dried, resuspended into a 100 μl of ultrapure sterile water, and treated with RNase at 37°C for 30 min. The DNA integrity was evaluated on a 1% agarose gel electrophoresis run, whereas the DNA purity was checked using Nanodrop™ spectrophotometer (Thermo Fisher Scientific). DNA sequencing was performed on a MinION Mk1b device (Oxford Nanopore Technologies, ONT, United Kingdom) using a R9.4.1 Flow Cell (ONT), after library preparation using the SQK-RBK004 Rapid Barcoding Kit (ONT). An aliquot of the same DNA sample was sequenced at Eurofins Genomics (Eurofins Genomics GmbH, Konstanz, Germany) with the genome sequencer Illumina NovaSeq 6000 S2 using the paired-end sequencing.
Genome Assembly
Illumina reads quality was evaluated using FastQC (Andrews, 2010), NovaSeq 6000 adapters were trimmed using Trimmomatic version 0.39 (Bolger et al., 2014), and low-quality reads were removed using Sickle (Joshi and Fass, 2011). To get the draft genome sequence, four different assemblers were used separately: SPAdes version 3.11.1 (Nurk et al., 2013), Minimap2 version 2.12-r849-dirty (Li, 2018) in combination with Miniasm version 0.3-r179 (Li, 2016), MaSuRCA version 3.4.2 (Zimin et al., 2013), and Canu version 2.1.1 (Koren et al., 2017). SPAdes was used in a first trial using only Illumina reads and in a second run for a hybrid assembly using both Illumina and Nanopore reads. A hybrid assembly was also performed using MaSuRCA, whereas Miniasm-Minimap2 and Canu used only Nanopore reads. For all the assemblers, default parameters were used, except for the expected genome size that was set at 40 Mb in Canu.
The obtained assemblies were further polished using Nanopolish version 0.11.1 (Loman et al., 2015), Racon version 1.3.3 (Vaser et al., 2017), or Pilon version 1.23 (Walker et al., 2014). The assembly quality statistics, before and after polishing, were evaluated using QUAST version 5.0.2 (Gurevich et al., 2013), while BUSCO version 5.beta.1 (Simão et al., 2015) was used to assess the assembly completeness, using hypocreales_db10 as ortholog lineage dataset, which consists of a set of 4,494 conserved profiles.
Genome Annotation
The draft genome assembled using Canu, chosen for downstream analysis, was structurally annotated following the de novo MAKER pipeline version 3.01.03 (Holt and Yandell, 2011), using the built-in RepeatModeler to mask repetitive elements, SNAP and AUGUSTUS for an ab initio gene prediction, and Est2Genome and Protein2Genome to further refine introns and exons boundaries using Exonerate and tRNAscan-SE to identify the genes related to the tRNA biosynthesis. Transcripts and proteins concatenated from four closely related Fusarium species (F. fujikuroi, F. graminearum, F. oxysporum f. sp. lycopersici, and F. verticillioides) were given as gene models, and the maximum intron size was set as 2,500. Functional annotation was performed using BLASTp and SwissProt as a database.
The same annotation pipeline was applied to annotate the genomes of F. culmorum, F. circinatum, F. oxysporum f. sp. koae 44, F. pseudograminearum CS3270, F. solani IlSc-1, F. tricinctum INRA104, F. tricinctum NRRL25481, F. tricinctum T6, F. verticilloides BRIP 53263, BRIP 53590, and all the F. avenaceum isolates, except for the already annotated Fa05001 used for the following comparative genomics analysis.
Comparative Genomics and Phylogenetic Analysis
The genome sequence of 27 others Fusarium species, together with their annotated proteins when available, were downloaded from the NCBI genome databases and used for phylogenetic and comparative genomics analysis (Table 1). Notably, the only F. lateritium genome sequence available in the NCBI database under the accession number GCA_014898835.1 refers to a strain isolated from a symptomatic elm tree in Pineville, Louisiana, United States (Kim et al., 2020).
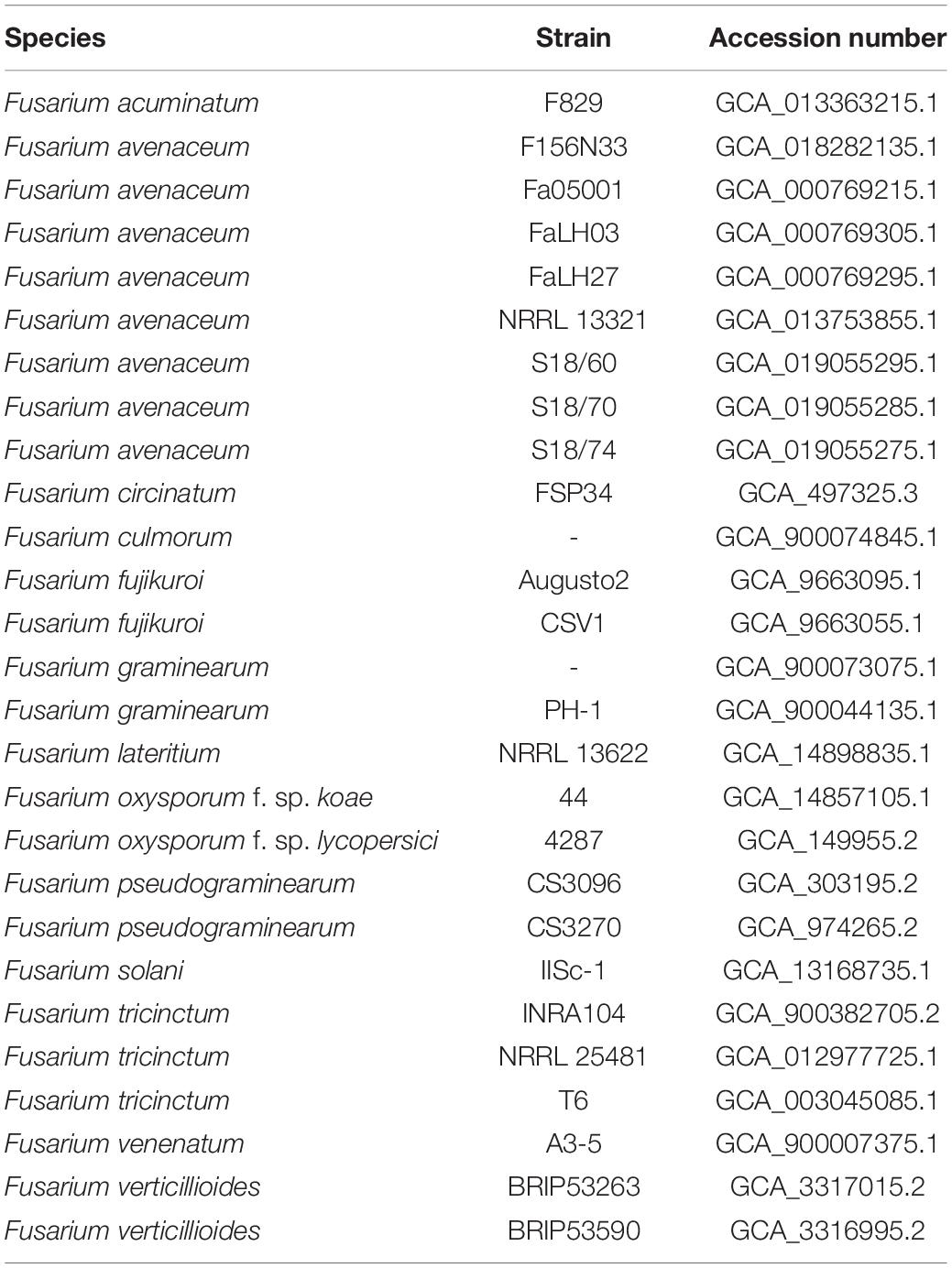
Table 1. List of Fusarium species used for pairwise genome comparisons and phylogenetic analysis.
For a phylogenetic analysis, the gene sequences of six housekeeping genes (EF-1α, RPB1, RPB2, beta tubulin, ITS, and LSU) were extracted from each genome and concatenated and aligned using MUSCLE (Edgar, 2004). The resulting alignment was used to build a maximum likelihood (ML) tree using raxmlHPC (Stamatakis, 2014) and visualized in a dendrogram using FigTree version 1.4.43. A second ML phylogenetic tree was built using raxmlHPC on the core genome SNPs identified during a pan genome analysis performed using Panseq, with the run mode set to pan, the fragment size at 500 nucleotides, the percentage of identity at 90%, and the core genome threshold to 28 genomes, in order to find out the sequences in common among all the strains (Laing et al., 2010). To better discriminate between isolates belonging to the F. tricinctum species complex (FTSC), a phylogenetic ML tree was built on the alignment of the concatenated sequence of the DNA-directed RNA polymerase II largest (RPB1) and second largest subunit (RPB2) nucleotide sequence, as previously applied by O’Donnell et al. (2013); Ponts et al. (2020), and Crous et al. (2021). MUSCLE and RAxML were used as previously described, and the 63 Fusarium isolates used in this analysis are reported in Supplementary Table 1.
Orthologous proteins were identified using OrthoFinder, and the results were used to build a species tree, visualized using FigTree as well (Emms and Kelly, 2019). The average nucleotide identity (ANI) analysis was performed using the pyani script and ANIb as algorithm for the alignment (Pritchard et al., 2016).
Characterization of Transcripts Involved in Pathogenesis
The putative secreted proteins involved in pathogenesis were identified using SignalP version 5.0b with the cutoff set ≥ 0.5 (Almagro Armenteros et al., 2019a). TargetP version 2.0 was used to identify signal peptide (SP), mitochondrial and chloroplast transit peptide (mTP and cTP, respectively), and potential cleavage sites (CS) (Almagro Armenteros et al., 2019b). Prediction of transmembrane proteins was performed using TMHMM version 2.0 (Krogh et al., 2001). Pathogen Host Interactions database (Urban et al., 2019) was used to find the similarity with pathogenicity and virulence-related genes, experimentally tested for roles in pathogenicity. Instead, Carbohydrate-Active Enzyme (CAZy) database (Lombard et al., 2014) was used to identify families of enzymes related to degradation, modification, and creation of glycosidic bonds, focusing on the Cell Wall Digestion Enzymes (CWDE).
The biosynthetic gene clusters (BGCs) were automatically searched and analyzed using AntiSMASH6 (Blin et al., 2021). The presence of the emerging mycotoxin enniatin was verified by blasting the available gene sequences from several Fusarium species (EF029060.1, NW_022158785.1, NW_022158526.1, KP000028.1, NC_030995.1, ENA| Z18755, NW_023502434.1, NW_023501408.1, and NW_023501343.1) (Fraeyman et al., 2017; Ponts et al., 2018).
Results
Strain Identification
A Fusarium sp. strain was isolated from symptomatic nuts collected from a hazelnut field located in the VT (Italy), which was being monitored in the framework of the PANTHEON project and named as Fusarium sp. isolate PT (Figures 1A,B). The morphological analysis performed to look at both macroscopic and microscopic characteristics revealed a mycelia color variable from white in the first 3–4 days to pale orange. Abundant medium-long, thin macroconidia with walls parallel for most of the spore length were observed, in the absence of microconidia and rare monophialides (Figures 1C,D). The characteristics found on the sample resembled the ones from the F. lateritium species when compared with The Fusarium Laboratory Manual (Leslie and Summerell, 2006). The sequenced ITS and the EF-1α region showed the highest identities (98.22 and 99.69% respectively) with F. lateritium sequences when blasted in the NCBI database (Supplementary Table 2).

Figure 1. Symptomatic hazelnuts and microscopic characteristics of the Fusarium sp. isolate PT. (A,B) The symptomatic fruits were characterized by a brown grayish necrotic spot/patch on the nut shell, bracts and less often on the petioles. (C) Macroconidia and (D) monophiliades of the isolated fungus visualized through optical microscope.
Pathogenicity Test
All fruits except the negative control developed symptoms similar to the one observed in the field (Figure 2). The Fusarium sp. isolate PT was consistently re-isolated from the inoculated nuts but not from the control fruits. The fungus re-isolated from diseased fruits showed the same morphology of the original isolate as well as the ITS, LSU, and β-tub, and EF-1α sequences were identical to those previously obtained, thus confirming Koch’s postulates.

Figure 2. Pathogen reinoculation. The isolated pathogen was reinoculated in healthy hazelnuts, which soon developed the same symptoms as the hazelnut collected in the field close to the Viterbo area (VT), confirming the Kock’s postulate.
Genome Assembly and Annotation
A clean and high molecular weight DNA was obtained, with both 260/230 nm and 260/280 nm ratios falling between 1.8 and 2 and was further sequenced using both ONT and Illumina technologies. ONT MinION run produced ∼701 k reads (1.92 Gbp; ∼73 × coverage), with a mean read length of 2,740 bp and an N50 of 4,588. Illumina NovaSeq 6000 S2 sequencing produced 2 × ∼2.5 M reads (2 Mbp × 385 Mbp; ∼20 × coverage).
After low-quality reads and adapters removal done by sickle and trimmomatic, respectively, the total number of the paired-end Illumina reads resulted to be 4.8 M in total, with an average length of 151 bp and a GC content of 47%. Instead, the average length of the Nanopore reads, affected by the starting DNA fragments material, resulted in an average length of 2,740 bp, with a GC content of 47%.
The reads were assembled following several pipelines and, thus, different algorithms whose results are described by QUAST statistics (Table 2). SPAdes draft genome derived either by only Illumina reads or by a hybrid assembly between Illumina and Nanopore reads, resulted to be the one with the lowest N50, the richest in undetermined bases N (171 and 48, respectively) and assembled in the highest number of contigs (70), and thus, it was discarded for further downstream analysis. The genome assembly derived from Miniasm-Minimap2 without any polishing yielded 70 contigs and 40.28 Mb of genome length with an N50 of 1.028 Mb that slightly increased to 1.029 Mb after five iterative polishing steps. MaSuRCA assembly resulted in 53 contigs arranged in a total length of 40.44 Mb and an N50 of 1.49 Mb. One step of Pilon polishing seemed to be unnecessary since the quality did not change much, proving that MaSuRCA itself with its POLCA polishing step is already enough to reach a good quality genome. Overall, Canu assembly resulted to be the one with the best statistics, with a total genome length of 40.51 Mb arranged in only 27 contigs.
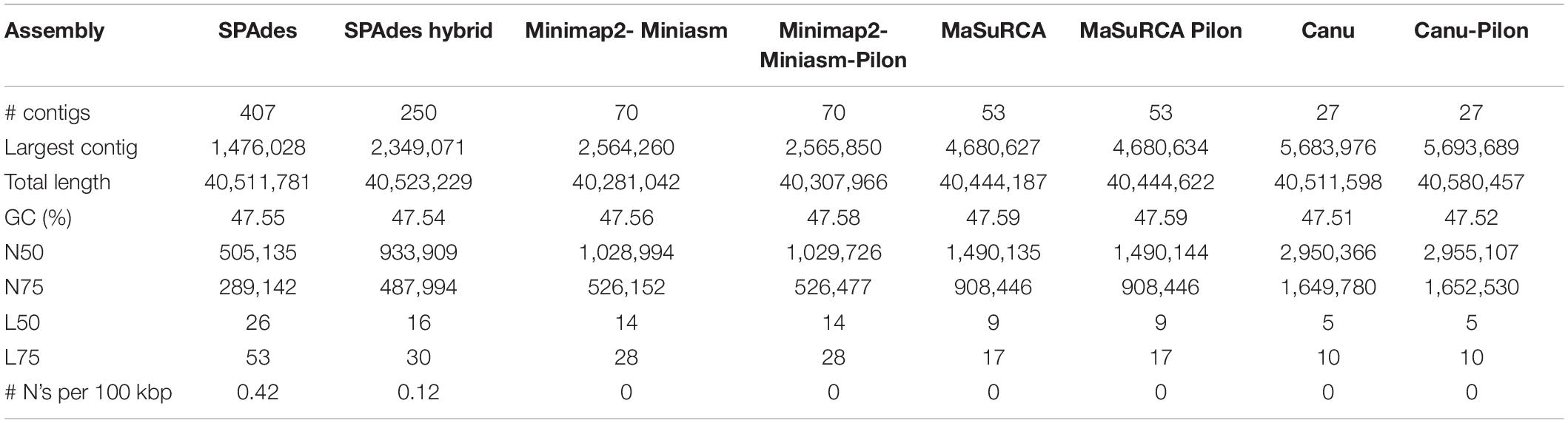
Table 2. Quast assembly statistics.
Then, the completeness of the three best quality assemblies was further evaluated by looking for the presence of 4,494 conserved ORF among the Hypocreales order (BUSCO). In fact, the polishing steps were needed to increase the quality and completeness of each of the three assemblies as well as to reduce the fragmented or missing BUSCO, leading to 99.6, 99.8, and 99.5% of completeness, respectively (Supplementary Dataset 1).
Based on the assembly statistics results, the draft genome assembled using Canu was chosen for downstream analysis. Its sequence was further verified by mapping the Illumina reads back on the 27 contigs used as reference to finally reach a high-quality consensus sequence (BWA version 0.7.17-r1188, samtools version 1.2, Burrows and Wheeler, 1994; Li et al., 2009). The genome was then annotated following MAKER pipeline and deposited on the NCBI genome database under the accession number JAHMRZ000000000.
Phylogenetic Analysis
The aligned concatenated sequences of EF-1α, RPB1, RPB2, beta tubulin, ITS, and LSU (around 7,100 nucleotides) of the Fusarium isolates under comparison showed clear differences perfectly represented by the ML tree in Figure 3A. Interestingly, our Fusarium sp. isolate PT clustered together with the FTSC and not directly with the other F. lateritium strain, as one could expect from the symptoms associated to the NGN. This clustering is due to the low sequence similarity, ranging from 82 to 86% of similarity, respectively, between the EF-1α, RPB1, RPB2, and beta-tubulin genes of the sequenced Fusarium sp. isolate PT and the F. lateritium, even though the ITS and LSU sequences were 100 and 98% identical (Supplementary Dataset 2). On the contrary, the sequence similarity of these genes with the ones from the F. tricinctum species complex were, indeed, higher but never reaching the 100% similarity (Supplementary Dataset 2).
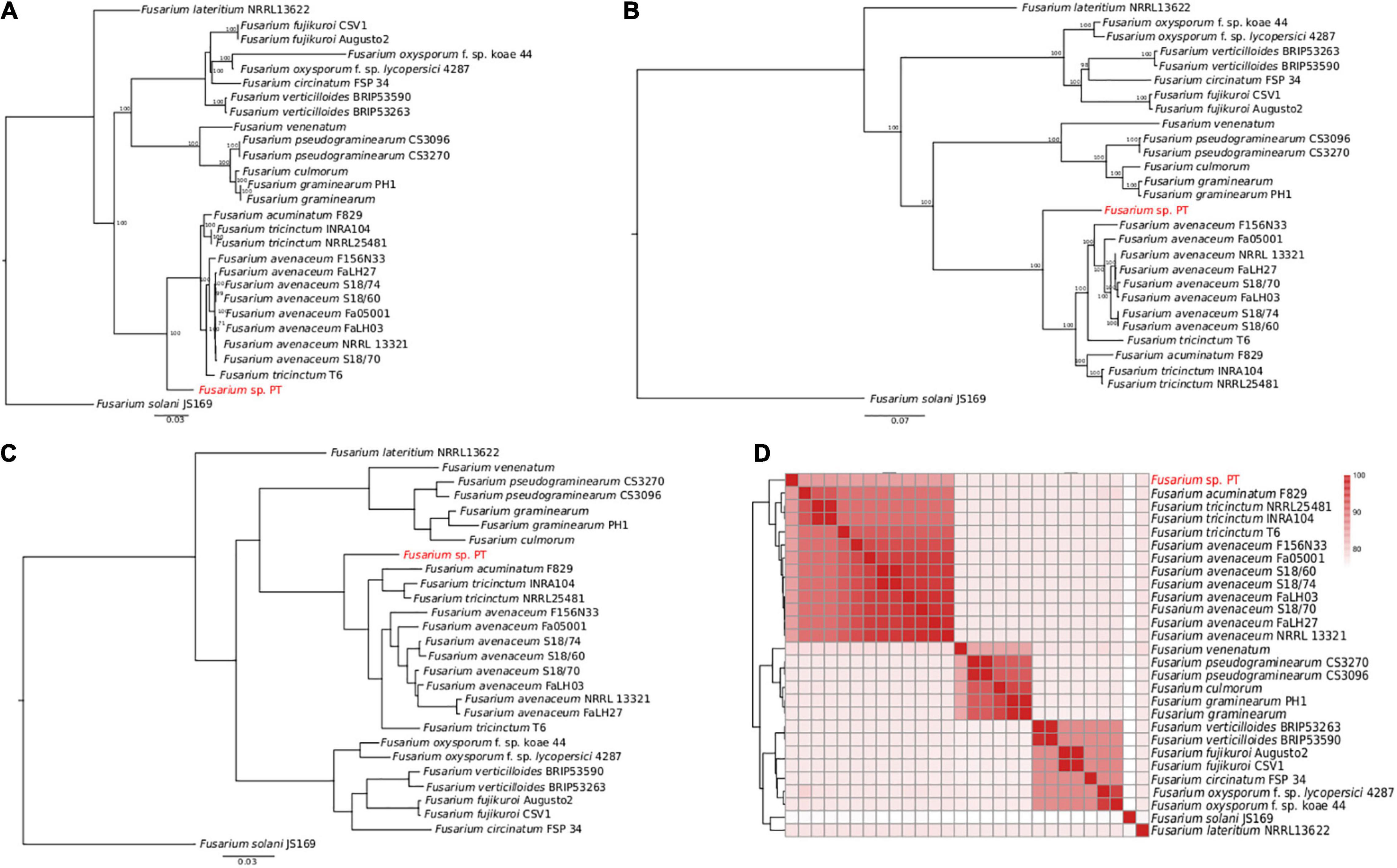
Figure 3. Phylogenetic analysis of 28 Fusarium strains. (A) The ML phylogenetic tree based on the alignment of the EF-1α, RPB1, RPB2, beta tubulin, ITS, and LSU concatenated nucleotide sequences. Only the bootstraps value higher than 70 are shown. (B) The ML phylogenetic tree based on the 4,319 core genome SNPs identified using Panseq. The number of bootstraps is indicated as well. (C) The ML phylogenetic tree of the orthologous proteins identified using OrthoFinder. In panels (A–C) Fusarium solani JS169 was used as outgroup. (D) Heatmap of the average nucleotide identity (ANI) performed using blastn showing the percentage of identify among the different Fusarium strains.
Accordingly, to further disentangle the phylogenetic relationship among the Fusarium strains, a ML tree was built on 4,319 SNPs found in the core genome alignment derived from Panseq. As shown in Figure 3B, the clustering of our Fusarium sp. isolate PT within the FTSC was confirmed. The same clusterization was obtained when the orthologous proteins were identified and a species tree was built using OrthoFinder (Figure 3C). The average nucleotide identity (ANI) performed using blastn gave an overall picture of the sequence identity between the Fusarium strains under comparison, as shown by the heatmap in Figure 3D, with the Fusarium sp. isolate PT again among the FTSC isolates.
Finally, a selection of 63 Fusarium isolates, belonging to the FTSC for which both RPB1 and RPB2 nucleotide sequences were available, was used to build an additional ML phylogenetic tree. As in the previous trees, the Fusarium sp. PT isolate constitutes an independent branch within the dendrogram (Supplementary Figure 1).
Characterization of Transcripts Involved in Pathogenesis
It is well-known that proteins are secreted in many Fusarium species during the colonizing stages (Ma et al., 2010), and for this reason, we went looking for the transcripts involved in pathogenesis.
In fact, 564 analogous genes involved in pathogenicity were identified from the PHI database. Among these, 24 genes were detected to be putative secreted proteins using SignalP. The 408 carbohydrate-active enzymes were detected, approximately 220 of which encoding glycoside hydrolases (GHs) and 79 genes encoding glycosyltransferases (Gts) (Table 3). Among the 1,288 proteins with a signal peptide, TargetP identified 525 proteins with a mitochondrial transit peptide, whereas 1,813 proteins resulted to have a possible cleavage site (Supplementary Figure 2). The predicted transmembrane helices found using TMHMM are reported in Supplementary Dataset 3.
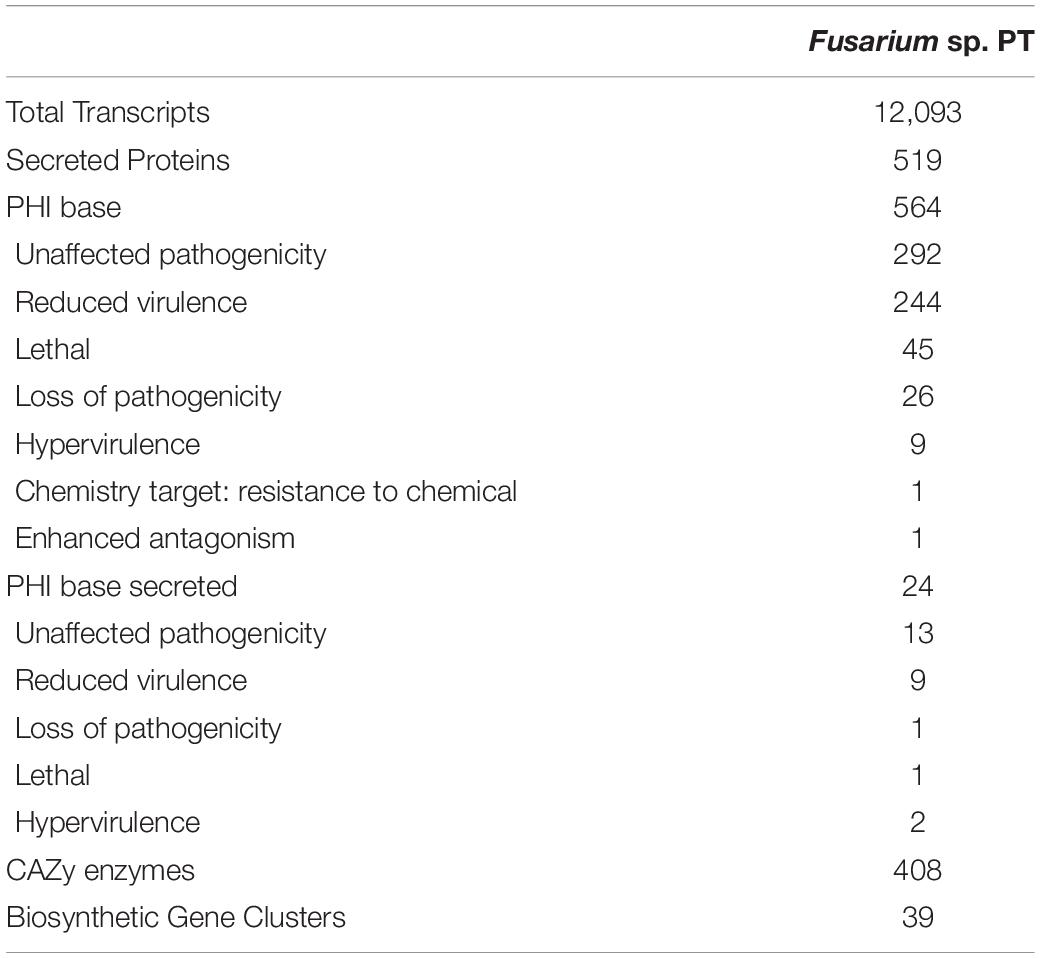
Table 3. Characterization of the transcripts involved in pathogenesis.
Seventeen biosynthetic gene clusters belonging to T1PKSs, NRPs, and terpene synthases (Tss) were found using antiSMASH 6. Among the T1PKSs gene cluster, the fujikurin A, B, C, and D were found with 83% of similarity, bikaverin with 42 or 57% of similarity, and fusarielin H and oxyjavanicin with 50% of similarity. Besides gibepyrone A with 40% of similarity, the ACT-Toxin II shared 100% identity with the sequence deposited in the database. The NRP gene cluster is represented mainly by ilicicolin H (50% of similarity) and chrysogine (83%), whereas BGCs belonging to bassianolide, beauvericin, and fusariodione A showed a similarity below 20%. Finally, the koraiol and α-acorenol terpenes showed 100% of similarity, whereas squalestatin S1 and gibberellin were of 40% similarity.
The complete sequence of the enniatin gene was also identified, sharing 85 and 86% of similarity with the gene deriving from F. scirpi and F. tricinctum strain INRA104, respectively (Supplementary Table 3).
Discussion
Fusarium is a wide fungal genus including numerous species, with an equally broad host range. Its classification has been traditionally based on morphological characters, such as asexual distinctive banana-shaped septate macroconidia (Leslie and Summerell, 2006). However, in the last decades, molecular approaches have made the species distinction more accurate, allowing the depiction of more than 300 phylogenetically distinct species (O’Donnell et al., 2004; Druzhinina et al., 2006). In most cases, the molecular analysis behind the species identification and the assignment of strains to definite species was, and still is, based on a multilocus sequence typing (MSLT) approach, meaning the comparison of complete or partial sequences of a bunch of housekeeping genes used, for example, in FUSARIUM-ID (Geiser et al., 2004; Park et al., 2010) and Fusarium MLST (O’Donnell et al., 2010). However, as already thoroughly discussed in several papers (see O’Donnell et al., 2015), even this approach during time has shown some limits. For instance, in Fusarium, ITS and LSU are often scarcely informative at species level and should be avoided, giving preference to EF1, RPB1, and RPB2.
In addition, it must be considered that the complexity of the genus and the reported criticalities in the selection of genes for species identification have sometimes led to misidentification of strains and, consequently, to species assignment in corresponding sequences when deposited in molecular databases.
It, therefore, seems clear why the new whole-genome sequencing (WGS) technologies are a powerful tool to solve these misunderstandings. Furthermore, they do not only allow accurate phylogenetic analysis but also provide the basic for a thorough understanding of molecular pathogenetic mechanisms involved in the plant-pathogen iteration, like the identification of the effector genes (Plissonneau et al., 2016; Möller and Stukenbrock, 2017). Knowledge of the infection pathways of the pathogen could allow to develop more effective control strategies. Thus, obtaining a high-quality genome as complete as possible, for those species who have not been sequenced yet, is an essential step to take.
In this study, we report the draft genome sequence of the strain Fusarium sp. isolate PT isolated from hazelnut in Central Italy, which initially was thought to be F. lateritium, due to the previous knowledge about the symptoms induced in the host and the morphology of the pathogen itself (Belisario and Santori, 2009; Santori et al., 2010; Vitale et al., 2011). In fact, its morphological traits observed under the optical microscope showed the typical banana-shaped septate macroconidia of F. lateritium (Leslie and Summerell, 2006). Furthermore, ITS and translation elongation factor 1-alpha sequencing corroborated this first hypothesis, guiding us toward a WGS with the aim to obtain a genome of this pathogen with the highest possible quality.
Accordingly, four different assembly approaches were applied, using both long reads obtained by ONT MinION and short reads by Illumina sequencing technologies, either in combination (hybrid assembly) or alone: (i) Miniasm-Minimap2, which use the overlap-layout-consensus (OLC); (ii) SPAdes uses the de Bruijn graph; (iii) MaSuRCA combines the de Bruijn graph with OLC, creating intermediate super-reads, further polished with POLCA; and (iv) Canu follows a MinHash Alignment Process (MHAP) based on k-mer weighting.
The advantage of using long reads compared with using only short reads has been extensively proved, especially for more complex genomes that are rich in transposons or tandem repeat (Goodwin et al., 2016; van Dijk et al., 2018). In addition, performing a hybrid assembly guarantee to take advantage, from one side, of the depth coverage and basecalling quality given by the Illumina reads and, on the other side, to increase the genome contiguity with the ONT reads (Chen et al., 2020). In fact, this method proved to be very successful in fungal genome assembly, either within the Fusarium world (Million et al., 2019; Degradi et al., 2021; Dvorianinova et al., 2021; Fan et al., 2021) or to extended species (Faino et al., 2015; Saud et al., 2021).
The same consideration applies to the assembly of the Fusarium sp. isolate in the study. In fact, the draft genome assembled by SPAdes using only Illumina reads gave the highest number of contigs but also undetermined nucleotides N, probably coming from repetitive regions or those gaps that Illumina reads could not fill. On the contrary, Canu, whose pipeline includes a de novo assembly from long reads followed by polishing with short reads, seemed to be the best strategy, also when compared with both MaSuRCA and Miniasm-Minimap2 hybrid assemblies. In fact, Canu produced less contigs (27) with the highest N50 (2,955,107bp) without Ns. In literature, good performance of Canu have been reported in F. musae (Degradi et al., 2021), F. oxysporum f. sp. capsici (Xingxing et al., 2021), and F. oxysporum f. sp. lini (Krasnov et al., 2020).
Accordingly, using the draft genome obtained using Canu for further analysis and annotation, different phylogenetic analysis surprising showed that the Fusarium sp. isolate PT is more closely related to F. tricinctum species complex than F. lateritium. These results were further supported by the species tree based on clustering of the orthologs proteins and on the dendrogram based on the ANI results.
Taken these results all together, it was once more demonstrated that species identification using only one or two housekeeping genes may led to wrong species assignment, as already shown for the ITS region in fungi such as Aspergillus, Fusarium, Penicillium, and Trichoderma (Raja et al., 2017).
In fact, polyphasic taxonomic approach, instead, gives a more robust classification of the species under examination, from the bacterial level to the more complicated Fusarium family (Das et al., 2014; Crous et al., 2021). In contrast, the still limited availability of WGSs in the databases may restrict the comparison range. But thanks to the drop in sequencing price and the constant increase in computational power, a WGS followed by comparative genomics analysis is becoming the best choice for a precise taxonomy identification. Moreover, each effort taken to increase the database availability contributes to fulfill gaps in fungal knowledge, especially for a variegated genus as the one of Fusarium.
At the same time, we did not go further in the attempt to assign a species name to the isolate in subject, leaving it at the status of undefined species. This agrees with the concept described by Summerell (2019) that the depiction of a new species must require the study of a number of strains with coherent genetic features that has to be sufficient to also describe the range of genetic variability within the new species.
From a strictly phytopathological point of view, this is the first report of a Fusarium isolate referable to the F. tricinctum species complex associated with the already known disease named NGN. In fact, to our knowledge, the only other report of Fusarium related to the tricinctum species complex associated with hazelnut comes from Iran and refers to a generic plant decline (Ghasemi and Davari, 2019). The NGN has been, instead, well studied and repeatedly attributed to the pathogenic action of F. lateritium (Belisario and Santori, 2009; Santori et al., 2010; Vitale et al., 2011), with a secondary role for Alternaria spp. Recently, the same disease was reported for the first time in La Araucanìa, Chile, and again different fungi were isolated from diseased fruit: Fusarium sp., even if the closest species according to ITS blast resulted F. sporotrichioides, but also Alternaria alternata, Diaporthe sp., Phomopsis sp., and Neofusicoccum sp. (Duran et al., 2020).
Several authors sustain the complexity of nut defects and rotting and the simultaneous occurrence of different pathogens in disease expression. Recently, Arciuolo et al. (2020), studying the fungal species associated with defective hazelnuts in Turkey, found that the prevalent fungi were Alternaria, Aspergillus, Botryosphaeria, Diaporthe, Fusarium, Penicillium, and Pestalotiopsis, proposing a major role for Diaporthe genus. In a previous paper, the species D. eres was demonstrated to be the main reason for the occurrence of brown spots on the kernel surface and of internal discoloration of nuts (Battilani et al., 2018).
In Oregon, Pscheidt et al. (2018), studying the fungi involved in kernel mold on hazelnut, found out that Penicillium spp., Aspergillus and Cladosporium spp., and D. rudis were frequently isolated, together with F. lateritium (identified by ITS and EF-1α).
All these studies demonstrate unequivocally that several fungi can concurrently invade and damage hazelnut generating various expressions of external and internal defects. Among them, Fusarium species undoubtedly have a key role in this type of disease. What remains to be assessed is the specific relevance of each of these species in the disease progression and which environmental factors influence the evolution of the disease. Investigating genomes as we did in this study, beside shedding light in the taxonomy of fungal species, particularly significant for the complex genus Fusarium, should allow to identify the genetic features of the pathogens involved in pathogenicity, thus representing milestones in understanding its evolution and eventually plan efficient control strategies to protect an important Italian crop as C. avellana.
Data Availability Statement
The Genome sequence can be found on the NCBI database genome under the accession number provided. The raw sequencing data are available under request.
Author Contributions
ST performed all the bioinformatics analysis, interpreted the results, prepared the figures, and wrote the manuscript in consultation with MD, LF, and AM. AG conceived and designed the study, collected the samples, and participated in the analysis. MD and CD performed the lab experiments. LF and MR provided the critical feedback and helped shape the research. VC and AM supervised the project and approved all the analysis. All authors contributed to the article and approved the submitted version.
Funding
This work has been supported by the European Commission under the Grant Agreement number 774571 (project PANTHEON, Precision Farming of Hazelnut Orchards).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We kindly thank Alessandro Infantino, Massimo Pilotti, and Antonio Matere of CREA-DC in Rome for the scientific and technical support in the microscopy analysis. All the bioinformatics calculations and analysis were performed at DAFNE HPC scientific computing center of the Università degli Studi della Tuscia.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2021.788584/full#supplementary-material
Supplementary Figure 1 | Phylogenetic tree of RPB1 and RPB2 concatenated sequences among FTSC strains. The nucleotide sequence of RPB1 and RPB2 genes of a selection of 63 strains belonging to the FTSC were concatenated, aligned, and used to build a ML tree using RAxML, from which only bootstraps higher than 60 are shown.
Supplementary Figure 2 | Results of TargetP analysis. Cumulative count of predicted proteins containing a signal peptide (SP), mitochondrial translocation signal (mTP), a possible cleavage site (CS) or other possible signal (chloroplast, extracellular, and “other” localization).
Supplementary Table 1 | List of 63 Fusarium isolates belonging to Fusarium tricinctum species complex (FTSC) used for pairwise comparison and phylogenetics analysis of the RPB1 and RPB2 gene sequences.
Supplementary Table 2 | List of the first 100 most significant sequence resulted from the BLASTn aligment of the ITS and the EF-1α region of the isolated strain.
Supplementary Table 3 | List of Fusarium strains showing identity with the enniatin gene of the Fusarium sp. isolate PT.
Footnotes
- ^ https://www.istat.it/
- ^ http://www.fao.org/faostat/
- ^ FigTree (http://tree.bio.ed.ac.uk/software/figtree)
References
Andrews, S. (2010). FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed September 30, 2021).
Arciuolo, R., Santos, C., Soares, C., Castello, G., Spigolon, N., Chiusa, G., et al. (2020). Molecular characterization of Diaporthe species associated with hazelnut defects. Front. Plant Sci. 11:1956. doi: 10.3389/fpls.2020.611655
Armenteros, J. J. A., Tsirigos, K. D., Sønderby, C. K., Petersen, T. N., Winther, O., Brunak, S., et al. (2019a). SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 37, 420–423. doi: 10.1038/s41587-019-0036-z
Armenteros, J. J. A., Salvatore, M., Emanuelsson, O., Winther, O., Heijne, G., von, et al. (2019b). Detecting sequence signals in targeting peptides using deep learning. Life Sci. Alliance 2:e201900429. doi: 10.26508/LSA.201900429
Battilani, P., Chiusa, G., Arciuolo, R., Somenzi, M., Fontana, M., Castello, G., et al. (2018). Diaporthe as the main cause of hazelnut defects in the Caucasus region. Phytopathol. Mediterr. 57, 320–333. doi: 10.14601/Phytopathol_Mediterr-22872
Belisario, A., and Santori, A. (2009). Gray necrosis of hazelnut fruit: a fungal disease causing fruit drop. Acta Hortic. 845, 501–506. doi: 10.17660/ActaHortic.2009.845.77
Blin, K., Shaw, S., Kloosterman, A. M., Charlop-Powers, Z., van Wezel, G. P., Medema, M. H., et al. (2021). antiSMASH 6.0: improving cluster detection and comparison capabilities. Nucleic Acids Res. 49, W29–W35. doi: 10.1093/NAR/GKAB335
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Burrows, M., and Wheeler, D. (1994). A Block-Sorting Lossless Data Compression Algorithm. Palo Alto, CA: Digital, Systems Research Center.
Chen, Z., Erickson, D. L., and Meng, J. (2020). Benchmarking hybrid assembly approaches for genomic analyses of bacterial pathogens using Illumina and Oxford Nanopore sequencing. BMC Genomics 21:631. doi: 10.1186/s12864-020-07041-8
Crous, P. W., Lombard, L., Sandoval-Denis, M., Seifert, K. A., Schroers, H. J., Chaverri, P., et al. (2021). Fusarium: more than a node or a foot-shaped basal cell. Stud. Mycol. 98:100116. doi: 10.1016/J.SIMYCO.2021.100116
Das, S., Dash, H. R., Mangwani, N., Chakraborty, J., and Kumari, S. (2014). Understanding molecular identification and polyphasic taxonomic approaches for genetic relatedness and phylogenetic relationships of microorganisms. J. Microbiol. Methods 103, 80–100. doi: 10.1016/j.mimet.2014.05.013
Degradi, M. L., Tava, M. V., Kunova, D. A., Cortesi, D. P., Saracchi, P. M., and Pasquali, D. M. (2021). Telomere to telomere genome assembly of Fusarium musae F31, causal agent of crown rot disease of banana. Mol. Plant Microbe Interact. [Epub ahead of print].
Druzhinina, I. S., Kopchinskiy, A. G., and Kubicek, C. P. (2006). The first 100 Trichoderma species characterized by molecular data. Mycoscience 47, 55–64. doi: 10.1007/S10267-006-0279-7
Duran, P., Barra, P. J., de la Luz Mora, M., Morina, F., Viscardi, Sh, and Meriño-Gergichevich, C. (2020). First report of fungal complex causing grey necrosis of hazelnut in Chile. New Dis. Rep. 42:7. doi: 10.5197/j.2044-0588.2020.042.007
Dvorianinova, E. M., Pushkova, E. N., Novakovskiy, R. O., Povkhova, L. V., Bolsheva, N. L., Kudryavtseva, L. P., et al. (2021). Nanopore and illumina genome sequencing of Fusarium oxysporum f. sp. lini strains of different virulence. Front. Genet. 12:662928. doi: 10.3389/FGENE.2021.662928
Edel-Hermann, V., Gautheron, N., Mounier, A., and Steinberg, C. (2015). Fusarium diversity in soil using a specific molecular approach and a cultural approach. J. Microbiol. Methods 111, 64–71. doi: 10.1016/j.mimet.2015.01.026
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Emms, D. M., and Kelly, S. (2019). OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 201, 1–14. doi: 10.1186/S13059-019-1832-Y
Faino, L., Seidl, M. F., Datema, E., Van Den Berg, G. C. M., Janssen, A., Wittenberg, A. H. J., et al. (2015). Single-molecule real-time sequencing combined with optical mapping yields completely finished fungal genome. mBio 6:e00936-15. doi: 10.1128/MBIO.00936-15
Fan, S., Wang, Q., Dai, J., Jiang, J., Hu, X., and Subbarao, K. V. (2021). The whole genome sequence of Fusarium redolens strain YP04, a pathogen that causes root rot of american ginseng. Phytopathology doi: 10.1094/PHYTO-03-21-0084-A [Epub ahead of print].
Fraeyman, S., Croubels, S., Devreese, M., and Antonissen, G. (2017). Emerging Fusarium and alternaria mycotoxins: occurrence, toxicity and toxicokinetics. Toxins 9:228. doi: 10.3390/TOXINS9070228
Geiser, D. M., del Mar Jiménez-Gasco, M., Kang, S., Makalowska, I., Veeraraghavan, N., Ward, T. J., et al. (2004). FUSARIUM-ID v. 1.0: a DNA sequence database for identifying Fusarium. Eur. J. Plant Pathol. 110, 473–479. doi: 10.1023/B:EJPP.0000032386.75915.a0
Ghasemi, S., and Davari, M. (2019). “Report of some Fusarium species associated with hazelnut decline in Ardabil,” in Proceedings of the 4th Iranian Mycological. Congress, 26–28 Aug., Sari Agricultural Sciences and Natural Resources, Iran.
Goodwin, S., McPherson, J. D., and McCombie, W. R. (2016). Coming of age: ten years of next-generation sequencing technologies. Nat. Rev. Genet. 17, 333–351. doi: 10.1038/nrg.2016.49
Graham, G. C., and Henry, R. J. (1997). “Preparation of fungal genomic DNA for PCR and RAPD analysis,” in Fingerprinting Methods Based on Arbitrarily Primed PCR, ed. Springer Lab Manual (Berlin: Springer Berlin Heidelberg), 29–34. doi: 10.1007/978-3-642-60441-6_5
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Holt, C., and Yandell, M. (2011). MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinform. 12:491. doi: 10.1186/1471-2105-12-491
Joshi, N., and Fass, J. (2011). Sickle: A Sliding-Window, Adaptive, Quality-Based Trimming Tool for FastQ Files (Version 1.33). Available online at: https://github.com/najoshi/sickle (accessed September 30, 2021).
Kim, H.-S., Lohmar, J. M., Busman, M., Brown, D. W., Naumann, T. A., Divon, H. H., et al. (2020). Identification and distribution of gene clusters required for synthesis of sphingolipid metabolism inhibitors in diverse species of the filamentous fungus Fusarium. BMC Genomics 21:712. doi: 10.1186/s12864-020-07135-3
Koren, S., Walenz, B. P., Berlin, K., Miller, J. R., Bergman, N. H., and Phillippy, A. M. (2017). Canu: scalable and accurate long-read assembly via adaptive k -mer weighting and repeat separation. Genome Res. 27, 722–736. doi: 10.1101/gr.215087.116
Krasnov, G. S., Pushkova, E. N., Novakovskiy, R. O., Kudryavtseva, L. P., Rozhmina, T. A., Dvorianinova, E. M., et al. (2020). High-quality genome assembly of Fusarium oxysporum f. sp. lini. Front. Genet. 11:959. doi: 10.3389/fgene.2020.00959
Krogh, A., Larsson, B., Von Heijne, G., and Sonnhammer, E. L. (2001). Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305, 567–580. doi: 10.1006/jmbi.2000.4315
Laing, C., Buchanan, C., Taboada, E. N., Zhang, Y., Kropinski, A., Villegas, A., et al. (2010). Pan-genome sequence analysis using Panseq: an online tool for the rapid analysis of core and accessory genomic regions. BMC Bioinformatics 11:461. doi: 10.1186/1471-2105-11-461
Leslie, J. F., and Summerell, B. A. (2006). The Fusarium Laboratory Manual. Hoboken, NJ: Blackwell Publishing Ltd.
Li, H. (2016). Minimap and miniasm: fast mapping and de novo assembly for noisy long sequences. Bioinformatics 32, 2103–2110. doi: 10.1093/bioinformatics/btw152
Li, H. (2018). Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100. doi: 10.1093/bioinformatics/bty191
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/BIOINFORMATICS/BTP352
Loman, N. J., Quick, J., and Simpson, J. T. (2015). A complete bacterial genome assembled de novo using only nanopore sequencing data. Nat. Methods 12, 733–735. doi: 10.1038/nmeth.3444
Lombard, V., Golaconda Ramulu, H., Drula, E., Coutinho, P. M., and Henrissat, B. (2014). The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42, D490–D495. doi: 10.1093/nar/gkt1178
Ma, L.-J., van der Does, H. C., Borkovich, K. A., Coleman, J. J., Daboussi, M.-J., Di Pietro, A., et al. (2010). Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature 464, 367–373. doi: 10.1038/nature08850
Million, C. R., Wijeratne, S., Cassone, B. J., Lee, S., Rouf Mian, M. A., McHale, L. K., et al. (2019). hybrid genome assembly of a major quantitative disease resistance locus in soybean toward Fusarium graminearum. Plant Genome 12:180102. doi: 10.3835/plantgenome2018.12.0102
Möller, M., and Stukenbrock, E. H. (2017). Evolution and genome architecture in fungal plant pathogens. Nat. Rev. Microbiol. 15, 756–771. doi: 10.1038/nrmicro.2017.76
Nurk, S., Bankevich, A., Antipov, D., Gurevich, A., Korobeynikov, A., Lapidus, A., et al. (2013). “Assembling genomes and mini-metagenomes from highly chimeric reads,” in Lecture Notes in Computer Science (including Subseries Lecture Notes in Artificial Intelligence and Lecture Notes in Bioinformatics, eds M. Deng, R. Jiang, F. Sun, and X. Zhang (Berlin: Springer), 158–170. doi: 10.1007/978-3-642-37195-0_13
O’Donnell, K., Rooney, A. P., Proctor, R. H., Brown, D. W., McCormick, S. P., Ward, T. J., et al. (2013). Phylogenetic analyses of RPB1 and RPB2 support a middle Cretaceous origin for a clade comprising all agriculturally and medically important fusaria. Fungal Genet. Biol. 52, 20–31. doi: 10.1016/j.fgb.2012.12.004
O’Donnell, K., Sutton, D. A., Rinaldi, M. G., Sarver, B. A. J., and Balajee, S. A. (2010). Internet-accessible DNA sequence database for identifying fusaria from human and animal infections. J. Clin. Microbiol. 48, 3708–3718. doi: 10.1128/JCM.00989-10
O’Donnell, K., Ward, T. J., Geiser, D. M., Kistler, H. C., and Aoki, T. (2004). Genealogical concordance between the mating type locus and seven other nuclear genes supports formal recognition of nine phylogenetically distinct species within the Fusarium graminearum clade. Fungal Genet. Biol. 41, 600–623. doi: 10.1016/j.fgb.2004.03.003
O’Donnell, K., Ward, T. J., Robert, V. A. R. G., Crous, P. W., Geiser, D. M., and Kang, S. (2015). DNA sequence-based identification of Fusarium: current status and future directions. Phytoparasitica 43, 583–595. doi: 10.1007/s12600-015-0484-z
Park, B., Park, J., Cheong, K.-C., Choi, J., Jung, K., Kim, D., et al. (2010). Cyber infrastructure for Fusarium: three integrated platforms supporting strain identification, phylogenetics, comparative genomics, and knowledge sharing. Nucleic Acids Res. 39, D640–D646.
Plissonneau, C., Stürchler, A., and Croll, D. (2016). The evolution of orphan regions in genomes of a fungal pathogen of wheat. mBio 7:e01231-16. doi: 10.1128/mBio.01231-16
Ponts, N., Gautier, C., Gouzy, J., Pinson-Gadais, L., Foulongne-Oriol, M., Ducos, C., et al. (2020). Evolution of Fusarium tricinctum and Fusarium avenaceum mitochondrial genomes is driven by mobility of introns and of a new type of palindromic microsatellite repeats. BMC Genomics 21:358. doi: 10.1186/s12864-020-6770-2
Ponts, N., Richard-Forget, F., Zhang, H., Barroso, G., and Zhao, C. (2018). Genome sequence of the emerging mycotoxin-producing filamentous fungus Fusarium tricinctum strain INRA104. Genome Announc 6, e509–e518. doi: 10.1128/GENOMEA.00509-18
Pritchard, L., Glover, R. H., Humphris, S., Elphinstone, J. G., and Toth, I. K. (2016). Genomics and taxonomy in diagnostics for food security: soft-rotting enterobacterial plant pathogens. Anal. Methods 8, 12–24. doi: 10.1039/C5AY02550H
Pscheidt, J. W., Heckert, S., Wiseman, M., and Jones, L. (2018). Fungi associated with and influence of moisture on development of kernel mold of hazelnut. Plant Dis. 103, 922–928. doi: 10.1094/PDIS-09-18-1520-RE
Raja, H. A., Miller, A. N., Pearce, C. J., and Oberlies, N. H. (2017). Fungal identification using molecular tools: a primer for the natural products research community. J. Nat. Prod. 80, 756–770. doi: 10.1021/acs.jnatprod.6b01085
Santori, A., Vitale, S., Luongo, L., and Belisario, A. (2010). First report of Fusarium lateritium as the agent of nut gray necrosis on hazelnut in Italy. Plant Dis. 94, 484–484. doi: 10.1094/PDIS-94-4-0484A
Saud, Z., Kortsinoglou, A. M., Kouvelis, V. N., and Butt, T. M. (2021). Telomere length de novo assembly of all 7 chromosomes and mitogenome sequencing of the model entomopathogenic fungus, Metarhizium brunneum, by means of a novel assembly pipeline. BMC Genomics 221:22. doi: 10.1186/S12864-021-07390-Y
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Summerell, B. A. (2019). Resolving Fusarium: current status of the genus. Annu. Rev. Phytopathol. 57, 323–339. doi: 10.1146/annurev-phyto-082718-100204
Urban, M., Cuzick, A., Seager, J., Wood, V., Rutherford, K., Venkatesh, S. Y., et al. (2019). PHI-base: the pathogen–host interactions database. Nucleic Acids Res. 48, D613–D620. doi: 10.1093/nar/gkz904
van Dijk, E. L., Jaszczyszyn, Y., Naquin, D., and Thermes, C. (2018). The third revolution in sequencing technology. Trends Genet. 34, 666–681. doi: 10.1016/j.tig.2018.05.008
Vaser, R., Sović, I., Nagarajan, N., and Šikić, M. (2017). Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 27, 737–746. doi: 10.1101/gr.214270.116
Vitale, S., Santori, A., Wajnberg, E., Castagnone-Sereno, P., Luongo, L., and Belisario, A. (2011). Morphological and molecular analysis of Fusarium lateritium, the cause of gray necrosis of hazelnut fruit in Italy. Phytopathology 101, 679–686. doi: 10.1094/PHYTO-04-10-0120
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9:e112963. doi: 10.1371/journal.pone.0112963
White, T. J., Bruns, T., Lee, S., and Taylor, J. (1990). “Amplification and direct sequencing of fungal ribosomal rna genes for phylogenetics,” in PCR Protocols : A Guide to Methods and Applications, eds M. A. I. D. H. Gelfand, J. J. Sninsky, and T. J. White (New York, NY: Academic Press), 315–322. doi: 10.1016/B978-0-12-372180-8.50042-1
Wollenweber, H. (1931). Fusarium on Walnut. Mitt. Deutsch. Dendrol. Gesellsch., XLIII (Jahrbuch). Berlin: Deutsche Dendrologische Gesellschaft.
Xingxing, P., Khan, R. A. A., Yan, L., Yuhong, Y., Bingyan, X., Zhenchuan, M., et al. (2021). Draft genome resource of Fusarium oxysporum f. sp. capsici, the infectious agent of pepper Fusarium wilt. Mol. Plant Microbe Interact. 34, 715–717. doi: 10.1094/MPMI-12-20-0355-A
Keywords: nut gray necrosis (NGN), Fusarium tricinctum species complex, hazelnut (Corylus avellana L.), genomics, hybrid assembly
Citation: Turco S, Grottoli A, Drais MI, De Spirito C, Faino L, Reverberi M, Cristofori V and Mazzaglia A (2021) Draft Genome Sequence of a New Fusarium Isolate Belonging to Fusarium tricinctum Species Complex Collected From Hazelnut in Central Italy. Front. Plant Sci. 12:788584. doi: 10.3389/fpls.2021.788584
Received: 02 October 2021; Accepted: 12 November 2021;
Published: 16 December 2021.
Edited by:
Marco Landi, University of Pisa, ItalyReviewed by:
Gerard Barroso, Université de Bordeaux, FranceJian Ling, Chinese Academy of Agricultural Sciences (CAAS), China
Copyright © 2021 Turco, Grottoli, Drais, De Spirito, Faino, Reverberi, Cristofori and Mazzaglia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Silvia Turco, silvia.turco@unitus.it; Angelo Mazzaglia, angmazza@unitus.it
†These authors have contributed equally to this work